 Liquid nitrogen (N2 at < −196 °C) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Nitrogen | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allotropes | see § Allotropes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Appearance | colorless gas, liquid or solid | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Standard atomic weight Ar°(N) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Nitrogen in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Group | group 15 (pnictogens) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Period | period 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Block | p-block | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electron configuration | [He] 2s2 2p3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Physical properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phase at STP | gas | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Melting point | (N2) 63.23[2] K (−209.86[2] °C, −345.75[2] °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Boiling point | (N2) 77.355 K (−195.795 °C, −320.431 °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density (at STP) | 1.2506 g/L[3] at 0 °C, 1013 mbar | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| when liquid (at b.p.) | 0.808 g/cm3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Triple point | 63.151 K, 12.52 kPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Critical point | 126.21 K, 3.39 MPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of fusion | (N2) 0.72 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of vaporisation | (N2) 5.57 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar heat capacity | (N2) 29.124 J/(mol·K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Vapour pressure
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxidation states | −3, −2, −1, 0,[4] +1, +2, +3, +4, +5 (a strongly acidic oxide) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 3.04 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ionisation energies |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Covalent radius | 71±1 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Van der Waals radius | 155 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Spectral lines of nitrogen | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Natural occurrence | primordial | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure | hexagonal
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Speed of sound | 353 m/s (gas, at 27 °C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal conductivity | 25.83×10−3 W/(m⋅K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Magnetic ordering | diamagnetic | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAS Number | 17778-88-0 7727-37-9 (N2) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| History | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Discovery | Daniel Rutherford (1772) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Named by | Jean-Antoine Chaptal (1790) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Main isotopes of nitrogen
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| references |

Nitrogen is the chemical element with the symbol N and atomic number 7. Nitrogen is a nonmetal and the lightest member of group 15 of the periodic table, often called the pnictogens. It is a common element in the universe, estimated at seventh in total abundance in the Milky Way and the Solar System. At standard temperature and pressure, two atoms of the element bond to form N2, a colorless and odorless diatomic gas. N2 forms about 78% of Earth’s atmosphere, making it the most abundant uncombined element. Nitrogen occurs in all organisms, primarily in amino acids (and thus proteins), in the nucleic acids (DNA and RNA) and in the energy transfer molecule adenosine triphosphate. The human body contains about 3% nitrogen by mass, the fourth most abundant element in the body after oxygen, carbon, and hydrogen. The nitrogen cycle describes the movement of the element from the air, into the biosphere and organic compounds, then back into the atmosphere.
Many industrially important compounds, such as ammonia, nitric acid, organic nitrates (propellants and explosives), and cyanides, contain nitrogen. The extremely strong triple bond in elemental nitrogen (N≡N), the second strongest bond in any diatomic molecule after carbon monoxide (CO),[5] dominates nitrogen chemistry. This causes difficulty for both organisms and industry in converting N2 into useful compounds, but at the same time it means that burning, exploding, or decomposing nitrogen compounds to form nitrogen gas releases large amounts of often useful energy. Synthetically produced ammonia and nitrates are key industrial fertilisers, and fertiliser nitrates are key pollutants in the eutrophication of water systems.
It was first discovered and isolated by Scottish physician Daniel Rutherford in 1772. Although Carl Wilhelm Scheele and Henry Cavendish had independently done so at about the same time, Rutherford is generally accorded the credit because his work was published first. The name nitrogène was suggested by French chemist Jean-Antoine-Claude Chaptal in 1790 when it was found that nitrogen was present in nitric acid and nitrates. Antoine Lavoisier suggested instead the name azote, from the Ancient Greek: ἀζωτικός «no life», as it is an asphyxiant gas; this name is used in several languages, including French, Italian, Russian, Romanian, Portuguese and Turkish, and appears in the English names of some nitrogen compounds such as hydrazine, azides and azo compounds.
Apart from its use in fertilisers and energy stores, nitrogen is a constituent of organic compounds as diverse as Kevlar used in high-strength fabric and cyanoacrylate used in superglue. Nitrogen is a constituent of every major pharmacological drug class, including antibiotics. Many drugs are mimics or prodrugs of natural nitrogen-containing signal molecules: for example, the organic nitrates nitroglycerin and nitroprusside control blood pressure by metabolizing into nitric oxide. Many notable nitrogen-containing drugs, such as the natural caffeine and morphine or the synthetic amphetamines, act on receptors of animal neurotransmitters.
History

Nitrogen compounds have a very long history, ammonium chloride having been known to Herodotus. They were well-known by the Middle Ages. Alchemists knew nitric acid as aqua fortis (strong water), as well as other nitrogen compounds such as ammonium salts and nitrate salts. The mixture of nitric and hydrochloric acids was known as aqua regia (royal water), celebrated for its ability to dissolve gold, the king of metals.[6]
The discovery of nitrogen is attributed to the Scottish physician Daniel Rutherford in 1772, who called it noxious air.[7][8] Though he did not recognise it as an entirely different chemical substance, he clearly distinguished it from Joseph Black’s «fixed air», or carbon dioxide.[9] The fact that there was a component of air that does not support combustion was clear to Rutherford, although he was not aware that it was an element. Nitrogen was also studied at about the same time by Carl Wilhelm Scheele,[10] Henry Cavendish,[11] and Joseph Priestley,[12] who referred to it as burnt air or phlogisticated air. French chemist Antoine Lavoisier referred to nitrogen gas as «mephitic air» or azote, from the Greek word άζωτικός (azotikos), «no life», due to it being asphyxiant.[13][14] In an atmosphere of pure nitrogen, animals died and flames were extinguished. Though Lavoisier’s name was not accepted in English since it was pointed out that all gases but oxygen are either asphyxiant or outright toxic, it is used in many languages (French, Italian, Portuguese, Polish, Russian, Albanian, Turkish, etc.; the German Stickstoff similarly refers to the same characteristic, viz. ersticken «to choke or suffocate») and still remains in English in the common names of many nitrogen compounds, such as hydrazine and compounds of the azide ion. Finally, it led to the name «pnictogens» for the group headed by nitrogen, from the Greek πνίγειν «to choke».[6]
The English word nitrogen (1794) entered the language from the French nitrogène, coined in 1790 by French chemist Jean-Antoine Chaptal (1756–1832),[15] from the French nitre (potassium nitrate, also called saltpeter) and the French suffix -gène, «producing», from the Greek -γενής (-genes, «begotten»). Chaptal’s meaning was that nitrogen is the essential part of nitric acid, which in turn was produced from nitre. In earlier times, niter had been confused with Egyptian «natron» (sodium carbonate) – called νίτρον (nitron) in Greek – which, despite the name, contained no nitrate.[16]
The earliest military, industrial, and agricultural applications of nitrogen compounds used saltpeter (sodium nitrate or potassium nitrate), most notably in gunpowder, and later as fertiliser. In 1910, Lord Rayleigh discovered that an electrical discharge in nitrogen gas produced «active nitrogen», a monatomic allotrope of nitrogen.[17] The «whirling cloud of brilliant yellow light» produced by his apparatus reacted with mercury to produce explosive mercury nitride.[18]
For a long time, sources of nitrogen compounds were limited. Natural sources originated either from biology or deposits of nitrates produced by atmospheric reactions. Nitrogen fixation by industrial processes like the Frank–Caro process (1895–1899) and Haber–Bosch process (1908–1913) eased this shortage of nitrogen compounds, to the extent that half of global food production (see Applications) now relies on synthetic nitrogen fertilisers.[19] At the same time, use of the Ostwald process (1902) to produce nitrates from industrial nitrogen fixation allowed the large-scale industrial production of nitrates as feedstock in the manufacture of explosives in the World Wars of the 20th century.[20][21]
Properties
Atomic

The shapes of the five orbitals occupied in nitrogen. The two colours show the phase or sign of the wave function in each region. From left to right: 1s, 2s (cutaway to show internal structure), 2px, 2py, 2pz.
A nitrogen atom has seven electrons. In the ground state, they are arranged in the electron configuration 1s2
2s2
2p1
x2p1
y2p1
z. It, therefore, has five valence electrons in the 2s and 2p orbitals, three of which (the p-electrons) are unpaired. It has one of the highest electronegativities among the elements (3.04 on the Pauling scale), exceeded only by chlorine (3.16), oxygen (3.44), and fluorine (3.98). (The light noble gases, helium, neon, and argon, would presumably also be more electronegative, and in fact are on the Allen scale.)[22] Following periodic trends, its single-bond covalent radius of 71 pm is smaller than those of boron (84 pm) and carbon (76 pm), while it is larger than those of oxygen (66 pm) and fluorine (57 pm). The nitride anion, N3−, is much larger at 146 pm, similar to that of the oxide (O2−: 140 pm) and fluoride (F−: 133 pm) anions.[22] The first three ionisation energies of nitrogen are 1.402, 2.856, and 4.577 MJ·mol−1, and the sum of the fourth and fifth is 16.920 MJ·mol−1. Due to these very high figures, nitrogen has no simple cationic chemistry.[23]
The lack of radial nodes in the 2p subshell is directly responsible for many of the anomalous properties of the first row of the p-block, especially in nitrogen, oxygen, and fluorine. The 2p subshell is very small and has a very similar radius to the 2s shell, facilitating orbital hybridisation. It also results in very large electrostatic forces of attraction between the nucleus and the valence electrons in the 2s and 2p shells, resulting in very high electronegativities. Hypervalency is almost unknown in the 2p elements for the same reason, because the high electronegativity makes it difficult for a small nitrogen atom to be a central atom in an electron-rich three-center four-electron bond since it would tend to attract the electrons strongly to itself. Thus, despite nitrogen’s position at the head of group 15 in the periodic table, its chemistry shows huge differences from that of its heavier congeners phosphorus, arsenic, antimony, and bismuth.[24]
Nitrogen may be usefully compared to its horizontal neighbours’ carbon and oxygen as well as its vertical neighbours in the pnictogen column, phosphorus, arsenic, antimony, and bismuth. Although each period 2 element from lithium to oxygen shows some similarities to the period 3 element in the next group (from magnesium to chlorine; these are known as diagonal relationships), their degree drops off abruptly past the boron–silicon pair. The similarities of nitrogen to sulfur are mostly limited to sulfur nitride ring compounds when both elements are the only ones present.[25]
Nitrogen does not share the proclivity of carbon for catenation. Like carbon, nitrogen tends to form ionic or metallic compounds with metals. Nitrogen forms an extensive series of nitrides with carbon, including those with chain-, graphitic-, and fullerenic-like structures.[26]
It resembles oxygen with its high electronegativity and concomitant capability for hydrogen bonding and the ability to form coordination complexes by donating its lone pairs of electrons. There are some parallels between the chemistry of ammonia NH3 and water H2O. For example, the capacity of both compounds to be protonated to give NH4+ and H3O+ or deprotonated to give NH2− and OH−, with all of these able to be isolated in solid compounds.[27]
Nitrogen shares with both its horizontal neighbours a preference for forming multiple bonds, typically with carbon, oxygen, or other nitrogen atoms, through pπ–pπ interactions.[25] Thus, for example, nitrogen occurs as diatomic molecules and therefore has very much lower melting (−210 °C) and boiling points (−196 °C) than the rest of its group, as the N2 molecules are only held together by weak van der Waals interactions and there are very few electrons available to create significant instantaneous dipoles. This is not possible for its vertical neighbours; thus, the nitrogen oxides, nitrites, nitrates, nitro-, nitroso-, azo-, and diazo-compounds, azides, cyanates, thiocyanates, and imino-derivatives find no echo with phosphorus, arsenic, antimony, or bismuth. By the same token, however, the complexity of the phosphorus oxoacids finds no echo with nitrogen.[25] Setting aside their differences, nitrogen and phosphorus form an extensive series of compounds with one another; these have chain, ring, and cage structures.[28]
Table of thermal and physical properties of nitrogen (N2) at atmospheric pressure:[29][30]
| Temperature (K) | Density (kg/m^3) | Specific heat (kJ/kg °C) | Dynamic viscosity (kg/m s) | Kinematic viscosity (m^2/s) | Thermal conductivity (W/m °C) | Thermal diffusivity (m^2/s) | Prandtl Number |
| 100 | 3.4388 | 1.07 | 6.88E-06 | 2.00E-06 | 0.00958 | 2.60E-06 | 0.768 |
| 150 | 2.2594 | 1.05 | 1.01E-05 | 4.45E-06 | 0.0139 | 5.86E-06 | 0.759 |
| 200 | 1.7108 | 1.0429 | 1.29E-05 | 7.57E-06 | 0.01824 | 1.02E-05 | 0.747 |
| 300 | 1.1421 | 1.0408 | 1.78E-05 | 1.56E-05 | 0.0262 | 2.20E-05 | 0.713 |
| 400 | 0.8538 | 1.0459 | 2.20E-05 | 2.57E-05 | 0.03335 | 3.73E-05 | 0.691 |
| 500 | 0.6824 | 1.0555 | 2.57E-05 | 3.77E-05 | 0.03984 | 5.53E-05 | 0.684 |
| 600 | 0.5687 | 1.0756 | 2.91E-05 | 5.12E-05 | 0.0458 | 7.49E-05 | 0.686 |
| 700 | 0.4934 | 1.0969 | 3.21E-05 | 6.67E-05 | 0.05123 | 9.47E-05 | 0.691 |
| 800 | 0.4277 | 1.1225 | 3.48E-05 | 8.15E-05 | 0.05609 | 1.17E-04 | 0.7 |
| 900 | 0.3796 | 1.1464 | 3.75E-05 | 9.11E-05 | 0.0607 | 1.39E-04 | 0.711 |
| 1000 | 0.3412 | 1.1677 | 4.00E-05 | 1.19E-04 | 0.06475 | 1.63E-04 | 0.724 |
| 1100 | 0.3108 | 1.1857 | 4.23E-05 | 1.36E-04 | 0.0685 | 1.86E-04 | 0.736 |
| 1200 | 0.2851 | 1.2037 | 4.45E-05 | 1.56E-04 | 0.07184 | 2.09E-04 | 0.748 |
| 1300 | 0.2591 | 1.219 | 4.66E-05 | 1.80E-04 | 0.081 | 2.56E-04 | 0.701 |
Isotopes

Table of nuclides (Segrè chart) from carbon to fluorine (including nitrogen). Orange indicates proton emission (nuclides outside the proton drip line); pink for positron emission (inverse beta decay); black for stable nuclides; blue for electron emission (beta decay); and violet for neutron emission (nuclides outside the neutron drip line). Proton number increases going up the vertical axis and neutron number going to the right on the horizontal axis.
Nitrogen has two stable isotopes: 14N and 15N. The first is much more common, making up 99.634% of natural nitrogen, and the second (which is slightly heavier) makes up the remaining 0.366%. This leads to an atomic weight of around 14.007 u.[22] Both of these stable isotopes are produced in the CNO cycle in stars, but 14N is more common as its neutron capture is the rate-limiting step. 14N is one of the five stable odd–odd nuclides (a nuclide having an odd number of protons and neutrons); the other four are 2H, 6Li, 10B, and 180mTa.[31]
The relative abundance of 14N and 15N is practically constant in the atmosphere but can vary elsewhere, due to natural isotopic fractionation from biological redox reactions and the evaporation of natural ammonia or nitric acid.[32] Biologically mediated reactions (e.g., assimilation, nitrification, and denitrification) strongly control nitrogen dynamics in the soil. These reactions typically result in 15N enrichment of the substrate and depletion of the product.[33]
The heavy isotope 15N was first discovered by S. M. Naudé in 1929, and soon after heavy isotopes of the neighbouring elements oxygen and carbon were discovered.[34] It presents one of the lowest thermal neutron capture cross-sections of all isotopes.[35] It is frequently used in nuclear magnetic resonance (NMR) spectroscopy to determine the structures of nitrogen-containing molecules, due to its fractional nuclear spin of one-half, which offers advantages for NMR such as narrower line width. 14N, though also theoretically usable, has an integer nuclear spin of one and thus has a quadrupole moment that leads to wider and less useful spectra.[22] 15N NMR nevertheless has complications not encountered in the more common 1H and 13C NMR spectroscopy. The low natural abundance of 15N (0.36%) significantly reduces sensitivity, a problem which is only exacerbated by its low gyromagnetic ratio, (only 10.14% that of 1H). As a result, the signal-to-noise ratio for 1H is about 300 times as much as that for 15N at the same magnetic field strength.[36] This may be somewhat alleviated by isotopic enrichment of 15N by chemical exchange or fractional distillation. 15N-enriched compounds have the advantage that under standard conditions, they do not undergo chemical exchange of their nitrogen atoms with atmospheric nitrogen, unlike compounds with labelled hydrogen, carbon, and oxygen isotopes that must be kept away from the atmosphere.[22] The 15N:14N ratio is commonly used in stable isotope analysis in the fields of geochemistry, hydrology, paleoclimatology and paleoceanography, where it is called δ15N.[37]
Of the ten other isotopes produced synthetically, ranging from 12N to 23N, 13N has a half-life of ten minutes and the remaining isotopes have half-lives on the order of seconds (16N and 17N) or milliseconds. No other nitrogen isotopes are possible as they would fall outside the nuclear drip lines, leaking out a proton or neutron.[38] Given the half-life difference, 13N is the most important nitrogen radioisotope, being relatively long-lived enough to use in positron emission tomography (PET), although its half-life is still short and thus it must be produced at the venue of the PET, for example in a cyclotron via proton bombardment of 16O producing 13N and an alpha particle.[39]
The radioisotope 16N is the dominant radionuclide in the coolant of pressurised water reactors or boiling water reactors during normal operation. It is produced from 16O (in water) via an (n,p) reaction, in which the 16O atom captures a neutron and expels a proton. It has a short half-life of about 7.1 s,[38] but during its decay back to 16O produces high-energy gamma radiation (5 to 7 MeV).[38][40] Because of this, access to the primary coolant piping in a pressurised water reactor must be restricted during reactor power operation.[40] It is a sensitive and immediate indicator of leaks from the primary coolant system to the secondary steam cycle and is the primary means of detection for such leaks.[40]
Chemistry and compounds
Allotropes

Molecular orbital diagram of dinitrogen molecule, N2. There are five bonding orbitals and two antibonding orbitals (marked with an asterisk; orbitals involving the inner 1s electrons not shown), giving a total bond order of three.
Atomic nitrogen, also known as active nitrogen, is highly reactive, being a triradical with three unpaired electrons. Free nitrogen atoms easily react with most elements to form nitrides, and even when two free nitrogen atoms collide to produce an excited N2 molecule, they may release so much energy on collision with even such stable molecules as carbon dioxide and water to cause homolytic fission into radicals such as CO and O or OH and H. Atomic nitrogen is prepared by passing an electric discharge through nitrogen gas at 0.1–2 mmHg, which produces atomic nitrogen along with a peach-yellow emission that fades slowly as an afterglow for several minutes even after the discharge terminates.[25]
Given the great reactivity of atomic nitrogen, elemental nitrogen usually occurs as molecular N2, dinitrogen. This molecule is a colourless, odourless, and tasteless diamagnetic gas at standard conditions: it melts at −210 °C and boils at −196 °C.[25] Dinitrogen is mostly unreactive at room temperature, but it will nevertheless react with lithium metal and some transition metal complexes. This is due to its bonding, which is unique among the diatomic elements at standard conditions in that it has an N≡N triple bond. Triple bonds have short bond lengths (in this case, 109.76 pm) and high dissociation energies (in this case, 945.41 kJ/mol), and are thus very strong, explaining dinitrogen’s low level of chemical reactivity.[25][41]
Other nitrogen oligomers and polymers may be possible. If they could be synthesised, they may have potential applications as materials with a very high energy density, that could be used as powerful propellants or explosives.[42] Under extremely high pressures (1.1 million atm) and high temperatures (2000 K), as produced in a diamond anvil cell, nitrogen polymerises into the single-bonded cubic gauche crystal structure. This structure is similar to that of diamond, and both have extremely strong covalent bonds, resulting in its nickname «nitrogen diamond».[43]

At atmospheric pressure, molecular nitrogen condenses (liquefies) at 77 K (−195.79 °C) and freezes at 63 K (−210.01 °C)[44] into the beta hexagonal close-packed crystal allotropic form. Below 35.4 K (−237.6 °C) nitrogen assumes the cubic crystal allotropic form (called the alpha phase).[45] Liquid nitrogen, a colourless fluid resembling water in appearance, but with 80.8% of the density (the density of liquid nitrogen at its boiling point is 0.808 g/mL), is a common cryogen.[46] Solid nitrogen has many crystalline modifications. It forms a significant dynamic surface coverage on Pluto[47] and outer moons of the Solar System such as Triton.[48] Even at the low temperatures of solid nitrogen it is fairly volatile and can sublime to form an atmosphere, or condense back into nitrogen frost. It is very weak and flows in the form of glaciers and on Triton geysers of nitrogen gas come from the polar ice cap region.[49]
Dinitrogen complexes

The first example of a dinitrogen complex to be discovered was [Ru(NH3)5(N2)]2+ (see figure at right), and soon many other such complexes were discovered. These complexes, in which a nitrogen molecule donates at least one lone pair of electrons to a central metal cation, illustrate how N2 might bind to the metal(s) in nitrogenase and the catalyst for the Haber process: these processes involving dinitrogen activation are vitally important in biology and in the production of fertilisers.[50][51]
Dinitrogen is able to coordinate to metals in five different ways. The more well-characterised ways are the end-on M←N≡N (η1) and M←N≡N→M (μ, bis-η1), in which the lone pairs on the nitrogen atoms are donated to the metal cation. The less well-characterised ways involve dinitrogen donating electron pairs from the triple bond, either as a bridging ligand to two metal cations (μ, bis-η2) or to just one (η2). The fifth and unique method involves triple-coordination as a bridging ligand, donating all three electron pairs from the triple bond (μ3-N2). A few complexes feature multiple N2 ligands and some feature N2 bonded in multiple ways. Since N2 is isoelectronic with carbon monoxide (CO) and acetylene (C2H2), the bonding in dinitrogen complexes is closely allied to that in carbonyl compounds, although N2 is a weaker σ-donor and π-acceptor than CO. Theoretical studies show that σ donation is a more important factor allowing the formation of the M–N bond than π back-donation, which mostly only weakens the N–N bond, and end-on (η1) donation is more readily accomplished than side-on (η2) donation.[25]
Today, dinitrogen complexes are known for almost all the transition metals, accounting for several hundred compounds. They are normally prepared by three methods:[25]
- Replacing labile ligands such as H2O, H−, or CO directly by nitrogen: these are often reversible reactions that proceed at mild conditions.
- Reducing metal complexes in the presence of a suitable co-ligand in excess under nitrogen gas. A common choice includes replacing chloride ligands with dimethylphenylphosphine (PMe2Ph) to make up for the smaller number of nitrogen ligands attached to the original chlorine ligands.
- Converting a ligand with N–N bonds, such as hydrazine or azide, directly into a dinitrogen ligand.
Occasionally the N≡N bond may be formed directly within a metal complex, for example by directly reacting coordinated ammonia (NH3) with nitrous acid (HNO2), but this is not generally applicable. Most dinitrogen complexes have colours within the range white-yellow-orange-red-brown; a few exceptions are known, such as the blue [{Ti(η5-C5H5)2}2-(N2)].[25]
Nitrides, azides, and nitrido complexes
Nitrogen bonds to almost all the elements in the periodic table except the first three noble gases, helium, neon, and argon, and some of the very short-lived elements after bismuth, creating an immense variety of binary compounds with varying properties and applications in which pentazenium tetraazidoborate has the highest nitrogen content.[25] Many binary compounds are known: with the exception of the nitrogen hydrides, oxides, and fluorides, these are typically called nitrides. Many stoichiometric phases are usually present for most elements (e.g. MnN, Mn6N5, Mn3N2, Mn2N, Mn4N, and MnxN for 9.2 < x < 25.3). They may be classified as «salt-like» (mostly ionic), covalent, «diamond-like», and metallic (or interstitial), although this classification has limitations generally stemming from the continuity of bonding types instead of the discrete and separate types that it implies. They are normally prepared by directly reacting a metal with nitrogen or ammonia (sometimes after heating), or by thermal decomposition of metal amides:[52]
- 3 Ca + N2 → Ca3N2
- 3 Mg + 2 NH3 → Mg3N2 + 3 H2 (at 900 °C)
- 3 Zn(NH2)2 → Zn3N2 + 4 NH3
Many variants on these processes are possible. The most ionic of these nitrides are those of the alkali metals and alkaline earth metals, Li3N (Na, K, Rb, and Cs do not form stable nitrides for steric reasons) and M3N2 (M = Be, Mg, Ca, Sr, Ba). These can formally be thought of as salts of the N3− anion, although charge separation is not actually complete even for these highly electropositive elements. However, the alkali metal azides NaN3 and KN3, featuring the linear N−
3 anion, are well-known, as are Sr(N3)2 and Ba(N3)2. Azides of the B-subgroup metals (those in groups 11 through 16) are much less ionic, have more complicated structures, and detonate readily when shocked.[52]

Mesomeric structures of borazine, (–BH–NH–)3
Many covalent binary nitrides are known. Examples include cyanogen ((CN)2), triphosphorus pentanitride (P3N5), disulfur dinitride (S2N2), and tetrasulfur tetranitride (S4N4). The essentially covalent silicon nitride (Si3N4) and germanium nitride (Ge3N4) are also known: silicon nitride, in particular, would make a promising ceramic if not for the difficulty of working with and sintering it. In particular, the group 13 nitrides, most of which are promising semiconductors, are isoelectronic with graphite, diamond, and silicon carbide and have similar structures: their bonding changes from covalent to partially ionic to metallic as the group is descended. In particular, since the B–N unit is isoelectronic to C–C, and carbon is essentially intermediate in size between boron and nitrogen, much of organic chemistry finds an echo in boron–nitrogen chemistry, such as in borazine («inorganic benzene»). Nevertheless, the analogy is not exact due to the ease of nucleophilic attack at boron due to its deficiency in electrons, which is not possible in a wholly carbon-containing ring.[52]
The largest category of nitrides are the interstitial nitrides of formulae MN, M2N, and M4N (although variable composition is perfectly possible), where the small nitrogen atoms are positioned in the gaps in a metallic cubic or hexagonal close-packed lattice. They are opaque, very hard, and chemically inert, melting only at very high temperatures (generally over 2500 °C). They have a metallic lustre and conduct electricity as do metals. They hydrolyse only very slowly to give ammonia or nitrogen.[52]
The nitride anion (N3−) is the strongest π donor known among ligands (the second-strongest is O2−). Nitrido complexes are generally made by the thermal decomposition of azides or by deprotonating ammonia, and they usually involve a terminal {≡N}3− group. The linear azide anion (N−
3), being isoelectronic with nitrous oxide, carbon dioxide, and cyanate, forms many coordination complexes. Further catenation is rare, although N4−
4 (isoelectronic with carbonate and nitrate) is known.[52]
Hydrides

Standard reduction potentials for nitrogen-containing species. Top diagram shows potentials at pH 0; bottom diagram shows potentials at pH 14.[53]
Industrially, ammonia (NH3) is the most important compound of nitrogen and is prepared in larger amounts than any other compound because it contributes significantly to the nutritional needs of terrestrial organisms by serving as a precursor to food and fertilisers. It is a colourless alkaline gas with a characteristic pungent smell. The presence of hydrogen bonding has very significant effects on ammonia, conferring on it its high melting (−78 °C) and boiling (−33 °C) points. As a liquid, it is a very good solvent with a high heat of vaporisation (enabling it to be used in vacuum flasks), that also has a low viscosity and electrical conductivity and high dielectric constant, and is less dense than water. However, the hydrogen bonding in NH3 is weaker than that in H2O due to the lower electronegativity of nitrogen compared to oxygen and the presence of only one lone pair in NH3 rather than two in H2O. It is a weak base in aqueous solution (pKb 4.74); its conjugate acid is ammonium, NH+
4. It can also act as an extremely weak acid, losing a proton to produce the amide anion, NH−
2. It thus undergoes self-dissociation, similar to water, to produce ammonium and amide. Ammonia burns in air or oxygen, though not readily, to produce nitrogen gas; it burns in fluorine with a greenish-yellow flame to give nitrogen trifluoride. Reactions with the other nonmetals are very complex and tend to lead to a mixture of products. Ammonia reacts on heating with metals to give nitrides.[54]
Many other binary nitrogen hydrides are known, but the most important are hydrazine (N2H4) and hydrogen azide (HN3). Although it is not a nitrogen hydride, hydroxylamine (NH2OH) is similar in properties and structure to ammonia and hydrazine as well. Hydrazine is a fuming, colourless liquid that smells similar to ammonia. Its physical properties are very similar to those of water (melting point 2.0 °C, boiling point 113.5 °C, density 1.00 g/cm3). Despite it being an endothermic compound, it is kinetically stable. It burns quickly and completely in air very exothermically to give nitrogen and water vapour. It is a very useful and versatile reducing agent and is a weaker base than ammonia.[55] It is also commonly used as a rocket fuel.[56]
Hydrazine is generally made by reaction of ammonia with alkaline sodium hypochlorite in the presence of gelatin or glue:[55]
- NH3 + OCl− → NH2Cl + OH−
- NH2Cl + NH3 → N
2H+
5 + Cl− (slow) - N
2H+
5 + OH− → N2H4 + H2O (fast)
(The attacks by hydroxide and ammonia may be reversed, thus passing through the intermediate NHCl− instead.) The reason for adding gelatin is that it removes metal ions such as Cu2+ that catalyses the destruction of hydrazine by reaction with monochloramine (NH2Cl) to produce ammonium chloride and nitrogen.[55]
Hydrogen azide (HN3) was first produced in 1890 by the oxidation of aqueous hydrazine by nitrous acid. It is very explosive and even dilute solutions can be dangerous. It has a disagreeable and irritating smell and is a potentially lethal (but not cumulative) poison. It may be considered the conjugate acid of the azide anion, and is similarly analogous to the hydrohalic acids.[55]
Halides and oxohalides

All four simple nitrogen trihalides are known. A few mixed halides and hydrohalides are known, but are mostly unstable; examples include NClF2, NCl2F, NBrF2, NF2H, NFH2, NCl2H, and NClH2.[57]
Five nitrogen fluorides are known. Nitrogen trifluoride (NF3, first prepared in 1928) is a colourless and odourless gas that is thermodynamically stable, and most readily produced by the electrolysis of molten ammonium fluoride dissolved in anhydrous hydrogen fluoride. Like carbon tetrafluoride, it is not at all reactive and is stable in water or dilute aqueous acids or alkalis. Only when heated does it act as a fluorinating agent, and it reacts with copper, arsenic, antimony, and bismuth on contact at high temperatures to give tetrafluorohydrazine (N2F4). The cations NF+
4 and N
2F+
3 are also known (the latter from reacting tetrafluorohydrazine with strong fluoride-acceptors such as arsenic pentafluoride), as is ONF3, which has aroused interest due to the short N–O distance implying partial double bonding and the highly polar and long N–F bond. Tetrafluorohydrazine, unlike hydrazine itself, can dissociate at room temperature and above to give the radical NF2•. Fluorine azide (FN3) is very explosive and thermally unstable. Dinitrogen difluoride (N2F2) exists as thermally interconvertible cis and trans isomers, and was first found as a product of the thermal decomposition of FN3.[57]
Nitrogen trichloride (NCl3) is a dense, volatile, and explosive liquid whose physical properties are similar to those of carbon tetrachloride, although one difference is that NCl3 is easily hydrolysed by water while CCl4 is not. It was first synthesised in 1811 by Pierre Louis Dulong, who lost three fingers and an eye to its explosive tendencies. As a dilute gas it is less dangerous and is thus used industrially to bleach and sterilise flour. Nitrogen tribromide (NBr3), first prepared in 1975, is a deep red, temperature-sensitive, volatile solid that is explosive even at −100 °C. Nitrogen triiodide (NI3) is still more unstable and was only prepared in 1990. Its adduct with ammonia, which was known earlier, is very shock-sensitive: it can be set off by the touch of a feather, shifting air currents, or even alpha particles.[57][58] For this reason, small amounts of nitrogen triiodide are sometimes synthesised as a demonstration to high school chemistry students or as an act of «chemical magic».[59] Chlorine azide (ClN3) and bromine azide (BrN3) are extremely sensitive and explosive.[60][61]
Two series of nitrogen oxohalides are known: the nitrosyl halides (XNO) and the nitryl halides (XNO2). The first is very reactive gases that can be made by directly halogenating nitrous oxide. Nitrosyl fluoride (NOF) is colourless and a vigorous fluorinating agent. Nitrosyl chloride (NOCl) behaves in much the same way and has often been used as an ionising solvent. Nitrosyl bromide (NOBr) is red. The reactions of the nitryl halides are mostly similar: nitryl fluoride (FNO2) and nitryl chloride (ClNO2) are likewise reactive gases and vigorous halogenating agents.[57]
Oxides

Nitrogen dioxide at −196 °C, 0 °C, 23 °C, 35 °C, and 50 °C. NO
2 converts to colourless dinitrogen tetroxide (N
2O
4) at low temperatures, and reverts to NO
2 at higher temperatures.
Nitrogen forms nine molecular oxides, some of which were the first gases to be identified: N2O (nitrous oxide), NO (nitric oxide), N2O3 (dinitrogen trioxide), NO2 (nitrogen dioxide), N2O4 (dinitrogen tetroxide), N2O5 (dinitrogen pentoxide), N4O (nitrosylazide),[62] and N(NO2)3 (trinitramide).[63] All are thermally unstable towards decomposition to their elements. One other possible oxide that has not yet been synthesised is oxatetrazole (N4O), an aromatic ring.[62]
Nitrous oxide (N2O), better known as laughing gas, is made by thermal decomposition of molten ammonium nitrate at 250 °C. This is a redox reaction and thus nitric oxide and nitrogen are also produced as byproducts. It is mostly used as a propellant and aerating agent for sprayed canned whipped cream, and was formerly commonly used as an anaesthetic. Despite appearances, it cannot be considered to be the anhydride of hyponitrous acid (H2N2O2) because that acid is not produced by the dissolution of nitrous oxide in water. It is rather unreactive (not reacting with the halogens, the alkali metals, or ozone at room temperature, although reactivity increases upon heating) and has the unsymmetrical structure N–N–O (N≡N+O−↔−N=N+=O): above 600 °C it dissociates by breaking the weaker N–O bond.[62]
Nitric oxide (NO) is the simplest stable molecule with an odd number of electrons. In mammals, including humans, it is an important cellular signaling molecule involved in many physiological and pathological processes.[64] It is formed by catalytic oxidation of ammonia. It is a colourless paramagnetic gas that, being thermodynamically unstable, decomposes to nitrogen and oxygen gas at 1100–1200 °C. Its bonding is similar to that in nitrogen, but one extra electron is added to a π* antibonding orbital and thus the bond order has been reduced to approximately 2.5; hence dimerisation to O=N–N=O is unfavourable except below the boiling point (where the cis isomer is more stable) because it does not actually increase the total bond order and because the unpaired electron is delocalised across the NO molecule, granting it stability. There is also evidence for the asymmetric red dimer O=N–O=N when nitric oxide is condensed with polar molecules. It reacts with oxygen to give brown nitrogen dioxide and with halogens to give nitrosyl halides. It also reacts with transition metal compounds to give nitrosyl complexes, most of which are deeply coloured.[62]
Blue dinitrogen trioxide (N2O3) is only available as a solid because it rapidly dissociates above its melting point to give nitric oxide, nitrogen dioxide (NO2), and dinitrogen tetroxide (N2O4). The latter two compounds are somewhat difficult to study individually because of the equilibrium between them, although sometimes dinitrogen tetroxide can react by heterolytic fission to nitrosonium and nitrate in a medium with high dielectric constant. Nitrogen dioxide is an acrid, corrosive brown gas. Both compounds may be easily prepared by decomposing a dry metal nitrate. Both react with water to form nitric acid. Dinitrogen tetroxide is very useful for the preparation of anhydrous metal nitrates and nitrato complexes, and it became the storable oxidiser of choice for many rockets in both the United States and USSR by the late 1950s. This is because it is a hypergolic propellant in combination with a hydrazine-based rocket fuel and can be easily stored since it is liquid at room temperature.[62]
The thermally unstable and very reactive dinitrogen pentoxide (N2O5) is the anhydride of nitric acid, and can be made from it by dehydration with phosphorus pentoxide. It is of interest for the preparation of explosives.[65] It is a deliquescent, colourless crystalline solid that is sensitive to light. In the solid state it is ionic with structure [NO2]+[NO3]−; as a gas and in solution it is molecular O2N–O–NO2. Hydration to nitric acid comes readily, as does analogous reaction with hydrogen peroxide giving peroxonitric acid (HOONO2). It is a violent oxidising agent. Gaseous dinitrogen pentoxide decomposes as follows:[62]
- N2O5 ⇌ NO2 + NO3 → NO2 + O2 + NO
- N2O5 + NO ⇌ 3 NO2
Oxoacids, oxoanions, and oxoacid salts
Many nitrogen oxoacids are known, though most of them are unstable as pure compounds and are known only as aqueous solutions or as salts. Hyponitrous acid (H2N2O2) is a weak diprotic acid with the structure HON=NOH (pKa1 6.9, pKa2 11.6). Acidic solutions are quite stable but above pH 4 base-catalysed decomposition occurs via [HONNO]− to nitrous oxide and the hydroxide anion. Hyponitrites (involving the N
2O2−
2 anion) are stable to reducing agents and more commonly act as reducing agents themselves. They are an intermediate step in the oxidation of ammonia to nitrite, which occurs in the nitrogen cycle. Hyponitrite can act as a bridging or chelating bidentate ligand.[66]
Nitrous acid (HNO2) is not known as a pure compound, but is a common component in gaseous equilibria and is an important aqueous reagent: its aqueous solutions may be made from acidifying cool aqueous nitrite (NO−
2, bent) solutions, although already at room temperature disproportionation to nitrate and nitric oxide is significant. It is a weak acid with pKa 3.35 at 18 °C. They may be titrimetrically analysed by their oxidation to nitrate by permanganate. They are readily reduced to nitrous oxide and nitric oxide by sulfur dioxide, to hyponitrous acid with tin(II), and to ammonia with hydrogen sulfide. Salts of hydrazinium N
2H+
5 react with nitrous acid to produce azides which further react to give nitrous oxide and nitrogen. Sodium nitrite is mildly toxic in concentrations above 100 mg/kg, but small amounts are often used to cure meat and as a preservative to avoid bacterial spoilage. It is also used to synthesise hydroxylamine and to diazotise primary aromatic amines as follows:[66]
- ArNH2 + HNO2 → [ArNN]Cl + 2 H2O
Nitrite is also a common ligand that can coordinate in five ways. The most common are nitro (bonded from the nitrogen) and nitrito (bonded from an oxygen). Nitro-nitrito isomerism is common, where the nitrito form is usually less stable.[66]

Fuming nitric acid contaminated with yellow nitrogen dioxide
Nitric acid (HNO3) is by far the most important and the most stable of the nitrogen oxoacids. It is one of the three most used acids (the other two being sulfuric acid and hydrochloric acid) and was first discovered by alchemists in the 13th century. It is made by the catalytic oxidation of ammonia to nitric oxide, which is oxidised to nitrogen dioxide, and then dissolved in water to give concentrated nitric acid. In the United States of America, over seven million tonnes of nitric acid are produced every year, most of which is used for nitrate production for fertilisers and explosives, among other uses. Anhydrous nitric acid may be made by distilling concentrated nitric acid with phosphorus pentoxide at low pressure in glass apparatus in the dark. It can only be made in the solid state, because upon melting it spontaneously decomposes to nitrogen dioxide, and liquid nitric acid undergoes self-ionisation to a larger extent than any other covalent liquid as follows:[66]
- 2 HNO3 ⇌ H
2NO+
3 + NO−
3 ⇌ H2O + [NO2]+ + [NO3]−
Two hydrates, HNO3·H2O and HNO3·3H2O, are known that can be crystallised. It is a strong acid and concentrated solutions are strong oxidising agents, though gold, platinum, rhodium, and iridium are immune to attack. A 3:1 mixture of concentrated hydrochloric acid and nitric acid, called aqua regia, is still stronger and successfully dissolves gold and platinum, because free chlorine and nitrosyl chloride are formed and chloride anions can form strong complexes. In concentrated sulfuric acid, nitric acid is protonated to form nitronium, which can act as an electrophile for aromatic nitration:[66]
- HNO3 + 2 H2SO4 ⇌ NO+
2 + H3O+ + 2 HSO−
4
The thermal stabilities of nitrates (involving the trigonal planar NO−
3 anion) depends on the basicity of the metal, and so do the products of decomposition (thermolysis), which can vary between the nitrite (for example, sodium), the oxide (potassium and lead), or even the metal itself (silver) depending on their relative stabilities. Nitrate is also a common ligand with many modes of coordination.[66]
Finally, although orthonitric acid (H3NO4), which would be analogous to orthophosphoric acid, does not exist, the tetrahedral orthonitrate anion NO3−
4 is known in its sodium and potassium salts:[66]
![{displaystyle {ce {NaNO3{}+Na2O->[{ce {Ag~crucible}}][{ce {300^{circ }C~for~7days}}]Na3NO4}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ec729bc88f520e08fdce8a013dec8ae601d28509)
These white crystalline salts are very sensitive to water vapour and carbon dioxide in the air:[66]
- Na3NO4 + H2O + CO2 → NaNO3 + NaOH + NaHCO3
Despite its limited chemistry, the orthonitrate anion is interesting from a structural point of view due to its regular tetrahedral shape and the short N–O bond lengths, implying significant polar character to the bonding.[66]
Organic nitrogen compounds
Nitrogen is one of the most important elements in organic chemistry. Many organic functional groups involve a carbon–nitrogen bond, such as amides (RCONR2), amines (R3N), imines (RC(=NR)R), imides (RCO)2NR, azides (RN3), azo compounds (RN2R), cyanates and isocyanates (ROCN or RCNO), nitrates (RONO2), nitriles and isonitriles (RCN or RNC), nitrites (RONO), nitro compounds (RNO2), nitroso compounds (RNO), oximes (RCR=NOH), and pyridine derivatives. C–N bonds are strongly polarised towards nitrogen. In these compounds, nitrogen is usually trivalent (though it can be tetravalent in quaternary ammonium salts, R4N+), with a lone pair that can confer basicity on the compound by being coordinated to a proton. This may be offset by other factors: for example, amides are not basic because the lone pair is delocalised into a double bond (though they may act as acids at very low pH, being protonated at the oxygen), and pyrrole is not acidic because the lone pair is delocalised as part of an aromatic ring.[67] The amount of nitrogen in a chemical substance can be determined by the Kjeldahl method.[68] In particular, nitrogen is an essential component of nucleic acids, amino acids and thus proteins, and the energy-carrying molecule adenosine triphosphate and is thus vital to all life on Earth.[67]
Occurrence

Nitrogen is the most common pure element in the earth, making up 78.1% of the volume of the atmosphere[6] (75.5% by mass), around 3.89 million gigatonnes. Despite this, it is not very abundant in Earth’s crust, making up somewhere around 19 parts per million of this, on par with niobium, gallium, and lithium. (This represents 300,000 to a million gigatonnes of nitrogen, depending on the mass of the crust.[69]) The only important nitrogen minerals are nitre (potassium nitrate, saltpetre) and soda nitre (sodium nitrate, Chilean saltpetre). However, these have not been an important source of nitrates since the 1920s, when the industrial synthesis of ammonia and nitric acid became common.[70]
Nitrogen compounds constantly interchange between the atmosphere and living organisms. Nitrogen must first be processed, or «fixed», into a plant-usable form, usually ammonia. Some nitrogen fixation is done by lightning strikes producing the nitrogen oxides, but most is done by diazotrophic bacteria through enzymes known as nitrogenases (although today industrial nitrogen fixation to ammonia is also significant). When the ammonia is taken up by plants, it is used to synthesise proteins. These plants are then digested by animals who use the nitrogen compounds to synthesise their proteins and excrete nitrogen-bearing waste. Finally, these organisms die and decompose, undergoing bacterial and environmental oxidation and denitrification, returning free dinitrogen to the atmosphere. Industrial nitrogen fixation by the Haber process is mostly used as fertiliser, although excess nitrogen–bearing waste, when leached, leads to eutrophication of freshwater and the creation of marine dead zones, as nitrogen-driven bacterial growth depletes water oxygen to the point that all higher organisms die. Furthermore, nitrous oxide, which is produced during denitrification, attacks the atmospheric ozone layer.[70]
Many saltwater fish manufacture large amounts of trimethylamine oxide to protect them from the high osmotic effects of their environment; conversion of this compound to dimethylamine is responsible for the early odour in unfresh saltwater fish.[71] In animals, free radical nitric oxide (derived from an amino acid), serves as an important regulatory molecule for circulation.[72]
Nitric oxide’s rapid reaction with water in animals results in the production of its metabolite nitrite. Animal metabolism of nitrogen in proteins, in general, results in the excretion of urea, while animal metabolism of nucleic acids results in the excretion of urea and uric acid. The characteristic odour of animal flesh decay is caused by the creation of long-chain, nitrogen-containing amines, such as putrescine and cadaverine, which are breakdown products of the amino acids ornithine and lysine, respectively, in decaying proteins.[73]
Production
Nitrogen gas is an industrial gas produced by the fractional distillation of liquid air, or by mechanical means using gaseous air (pressurised reverse osmosis membrane or pressure swing adsorption). Nitrogen gas generators using membranes or pressure swing adsorption (PSA) are typically more cost and energy efficient than bulk-delivered nitrogen.[74] Commercial nitrogen is often a byproduct of air-processing for industrial concentration of oxygen for steelmaking and other purposes. When supplied compressed in cylinders it is often called OFN (oxygen-free nitrogen).[75] Commercial-grade nitrogen already contains at most 20 ppm oxygen, and specially purified grades containing at most 2 ppm oxygen and 10 ppm argon are also available.[76]
In a chemical laboratory, it is prepared by treating an aqueous solution of ammonium chloride with sodium nitrite.[77]
- NH4Cl + NaNO2 → N2 + NaCl + 2 H2O
Small amounts of the impurities NO and HNO3 are also formed in this reaction. The impurities can be removed by passing the gas through aqueous sulfuric acid containing potassium dichromate.[77] Very pure nitrogen can be prepared by the thermal decomposition of barium azide or sodium azide.[78]
- 2 NaN3 → 2 Na + 3 N2
Applications
Gas
The applications of nitrogen compounds are naturally extremely widely varied due to the huge size of this class: hence, only applications of pure nitrogen itself will be considered here. Two-thirds (2/3) of nitrogen produced by industry is sold as gas and the remaining one-third (1/3) as a liquid.
The gas is mostly used as a low reactivity safe atmosphere wherever the oxygen in the air would pose a fire, explosion, or oxidising hazard. Some examples include:[76]
- As a modified atmosphere, pure or mixed with carbon dioxide, to nitrogenate and preserve the freshness of packaged or bulk foods (by delaying rancidity and other forms of oxidative damage). Pure nitrogen as food additive is labeled in the European Union with the E number E941.[79]
- In incandescent light bulbs as an inexpensive alternative to argon.[80]
- In fire suppression systems for Information technology (IT) equipment.[76]
- In the manufacture of stainless steel.[81]
- In the case-hardening of steel by nitriding.[82]
- In some aircraft fuel systems to reduce fire hazard (see inerting system).
- To inflate race car and aircraft tires,[83] reducing the problems of inconsistent expansion and contraction caused by moisture and oxygen in natural air.[76]
Nitrogen is commonly used during sample preparation in chemical analysis. It is used to concentrate and reduce the volume of liquid samples. Directing a pressurised stream of nitrogen gas perpendicular to the surface of the liquid causes the solvent to evaporate while leaving the solute(s) and un-evaporated solvent behind.[84]
Nitrogen can be used as a replacement, or in combination with, carbon dioxide to pressurise kegs of some beers, particularly stouts and British ales, due to the smaller bubbles it produces, which makes the dispensed beer smoother and headier.[85] A pressure-sensitive nitrogen capsule known commonly as a «widget» allows nitrogen-charged beers to be packaged in cans and bottles.[86][87] Nitrogen tanks are also replacing carbon dioxide as the main power source for paintball guns. Nitrogen must be kept at a higher pressure than CO2, making N2 tanks heavier and more expensive.[88]
Equipment
Some construction equipment uses pressurized nitrogen gas to help hydraulic system to provide extra power to devices such as hydraulic hammer. Nitrogen gas, formed from the decomposition of sodium azide, is used for the inflation of airbags.[89]
Execution
As nitrogen is an asphyxiant gas, some jurisdictions have considered asphyxiation by inhalation of pure nitrogen as a means of capital punishment (as a substitute for lethal injection).[90][91][92]
However, as of 2020, no executions using nitrogen gas have yet been carried out by any jurisdiction, and at least one jurisdiction (Oklahoma) which had considered nitrogen asphyxiation as an execution protocol had abandoned the effort.[93]
Liquid
Air balloon submerged in liquid nitrogen
Liquid nitrogen is a cryogenic liquid which looks like water. When insulated in proper containers such as dewar flasks, it can be transported and stored with a low rate of evaporative loss.[94]

A container vehicle carrying liquid nitrogen.
Like dry ice, the main use of liquid nitrogen is for cooling to low temperatures. It is used in the cryopreservation of biological materials such as blood and reproductive cells (sperm and eggs). It is used in cryotherapy to remove cysts and warts on the skin by freezing them.[95] It is used in laboratory cold traps, and in cryopumps to obtain lower pressures in vacuum pumped systems. It is used to cool heat-sensitive electronics such as infrared detectors and X-ray detectors. Other uses include freeze-grinding and machining materials that are soft or rubbery at room temperature, shrink-fitting and assembling engineering components, and more generally to attain very low temperatures where necessary. Because of its low cost, liquid nitrogen is often used for cooling even when such low temperatures are not strictly necessary, such as refrigeration of food, freeze-branding livestock, freezing pipes to halt flow when valves are not present, and consolidating unstable soil by freezing whenever excavation is going on underneath.[76]
Safety
Gas
Although nitrogen is non-toxic, when released into an enclosed space it can displace oxygen, and therefore presents an asphyxiation hazard. This may happen with few warning symptoms, since the human carotid body is a relatively poor and slow low-oxygen (hypoxia) sensing system.[96] An example occurred shortly before the launch of the first Space Shuttle mission on March 19, 1981, when two technicians died from asphyxiation after they walked into a space located in the Space Shuttle’s mobile launcher platform that was pressurised with pure nitrogen as a precaution against fire.[97]
When inhaled at high partial pressures (more than about 4 bar, encountered at depths below about 30 m in scuba diving), nitrogen is an anesthetic agent, causing nitrogen narcosis, a temporary state of mental impairment similar to nitrous oxide intoxication.[98][99]
Nitrogen dissolves in the blood and body fats. Rapid decompression (as when divers ascend too quickly or astronauts decompress too quickly from cabin pressure to spacesuit pressure) can lead to a potentially fatal condition called decompression sickness (formerly known as caisson sickness or the bends), when nitrogen bubbles form in the bloodstream, nerves, joints, and other sensitive or vital areas.[100][101] Bubbles from other «inert» gases (gases other than carbon dioxide and oxygen) cause the same effects, so replacement of nitrogen in breathing gases may prevent nitrogen narcosis, but does not prevent decompression sickness.[102]
Liquid
As a cryogenic liquid, liquid nitrogen can be dangerous by causing cold burns on contact, although the Leidenfrost effect provides protection for very short exposure (about one second).[103] Ingestion of liquid nitrogen can cause severe internal damage. For example, in 2012, a young woman in England had to have her stomach removed after ingesting a cocktail made with liquid nitrogen.[104]
Because the liquid-to-gas expansion ratio of nitrogen is 1:694 at 20 °C, a tremendous amount of force can be generated if liquid nitrogen is rapidly vaporised in an enclosed space. In an incident on January 12, 2006, at Texas A&M University, the pressure-relief devices of a tank of liquid nitrogen were malfunctioning and later sealed. As a result of the subsequent pressure buildup, the tank failed catastrophically. The force of the explosion was sufficient to propel the tank through the ceiling immediately above it, shatter a reinforced concrete beam immediately below it, and blow the walls of the laboratory 0.1–0.2 m off their foundations.[105]
Liquid nitrogen readily evaporates to form gaseous nitrogen, and hence the precautions associated with gaseous nitrogen also apply to liquid nitrogen.[106][107][108] For example, oxygen sensors are sometimes used as a safety precaution when working with liquid nitrogen to alert workers of gas spills into a confined space.[109]
Vessels containing liquid nitrogen can condense oxygen from air. The liquid in such a vessel becomes increasingly enriched in oxygen (boiling point −183 °C, higher than that of nitrogen) as the nitrogen evaporates, and can cause violent oxidation of organic material.[110]
Oxygen deficiency monitors
Oxygen deficiency monitors are used to measure levels of oxygen in confined spaces and any place where nitrogen gas or liquid are stored or used. In the event of a nitrogen leak, and a decrease in oxygen to a pre-set alarm level, an oxygen deficiency monitor can be programmed to set off audible and visual alarms, thereby providing notification of the possible impending danger. Most commonly the oxygen range to alert personnel is when oxygen levels get below 19.5%. OSHA specifies that a hazardous atmosphere may include one where the oxygen concentration is below 19.5% or above 23.5%.[111]
Oxygen deficiency monitors can either be fixed, mounted to the wall and hard-wired into the building’s power supply or simply plugged into a power outlet, or a portable hand-held or wearable monitor.
See also
- Reactive nitrogen species
- Soil gas
References
- ^ «Standard Atomic Weights: Nitrogen». CIAAW. 2009.
- ^ a b c Lide, David R. (1990–1991). CRC Handbook of Physics and Chemistry (71st ed.). Boca Raton, Ann Arbor, Boston: CRC Press, inc. pp. 4-22 (one page).
- ^ «Gases — Density». The Engineering Toolbox. Retrieved 27 January 2019.
- ^ Tetrazoles contain a pair of double-bonded nitrogen atoms with oxidation state 0 in the ring. A Synthesis of the parent 1H-tetrazole, CH2N4 (two atoms N(0)) is given in Ronald A. Henry and William G. Finnegan, «An Improved Procedure for the Deamination of 5-Aminotetrazole», _J. Am. Chem. Soc._ (1954), 76, 1, 290–291,
https://doi.org/10.1021/ja01630a086. - ^ Common Bond Energies (D) and Bond Lengths (r) Archived 2010-05-15 at the Wayback Machine. wiredchemist.com
- ^ a b c Greenwood and Earnshaw, pp. 406–07
- ^ Rutherford, Daniel (1772) «Dissertatio Inauguralis de aere fixo, aut mephitico Archived 2020-08-06 at the Wayback Machine» (Inaugural dissertation on the air [called] fixed or mephitic), M.D. dissertation, University of Edinburgh, Scotland. English translation: Dobbin, Leonard (1935). «Daniel Rutherford’s inaugural dissertation». Journal of Chemical Education. 12 (8): 370–75. Bibcode:1935JChEd..12..370D. doi:10.1021/ed012p370.
- ^ Weeks, Mary Elvira (1932). «The discovery of the elements. IV. Three important gases». Journal of Chemical Education. 9 (2): 215. Bibcode:1932JChEd…9..215W. doi:10.1021/ed009p215.
- ^ Aaron J. Ihde, The Development of Modern Chemistry, New York 1964.
- ^ Carl Wilhelm Scheele, Chemische Abhandlung von der Luft und dem Feuer [Chemical treatise on air and fire] (Upsala, Sweden: Magnus Swederus, 1777; and Leipzig, (Germany): Siegfried Lebrecht Crusius, 1777). In the section titled «Die Luft muß aus elastischen Flüßigkeiten von zweyerley Art, zusammengesetzet seyn.» (The air must be composed of elastic fluids of two sorts), pp. 6–14, Scheele presents the results of eight experiments in which air was reacted with various substances. He concluded (p. 13): «So viel sehe ich aus angeführten Versuchen, daß die Luft aus 2 von einander unterschiedenen Flußigkeiten bestehe, von welchen die eine die Eigenschaft das Phlogiston anzuziehen gar nicht äussere, die andere aber zur solchen Attraction eigentlich aufgeleget ist und welche zwischen dem 3:ten und 4:ten Theil von der ganzen Luftmasse aus machet.» (So I see [this] much from the experiments [that were] conducted: that the air consists of two fluids [that] differ from one another, of which the one doesn’t express at all the property of attracting phlogiston; the other, however, is capable of such attraction and which makes up between 1/3 and 1/4 part of the entire mass of the air.)
- ^ Priestley, Joseph (1772). «Observations on different kinds of air». Philosophical Transactions of the Royal Society of London. 62: 147–256. doi:10.1098/rstl.1772.0021. S2CID 186210131. ; see p. 225. Archived 2016-09-03 at the Wayback Machine
- ^ Priestley, Joseph (1772). «Observations on different kinds of air». Philosophical Transactions of the Royal Society of London. 62: 147–256. doi:10.1098/rstl.1772.0021. S2CID 186210131. ; see: «VII. Of air infected with the fumes of burning charcoal.» pp. 225–28. Archived 2016-09-03 at the Wayback Machine
- ^ Lavoisier, Antoine with Robert Kerr, trans., Elements of Chemistry, 4th ed. (Edinburgh, Scotland: William Creech, 1799), pp. 85–86. [p. 85]: Archived 2020-08-06 at the Wayback Machine «In reflecting upon the circumstances of this experiment, we readily perceive; that the mercury, during its calcination [i.e., roasting in air], absorbs the salubrious and respirable part of the air, or, to speak more strictly, the base of this respirable part; that the remaining air is a species of mephitis [i.e., a poisonous gas emitted from the earth], incapable of supporting combustion or respiration; … » [p. 86]: Archived 2020-08-06 at the Wayback Machine «I shall afterwards shew, that at least in our climate, the atmospheric air is composed of respirable and mephitic airs, in the proportion of 27 and 73; … «
- ^ Lavoisier, Antoine with Robert Kerr, trans., Elements of Chemistry, 4th ed. (Edinburgh, Scotland: William Creech, 1799), p. 101: «The chemical properties of the noxious portion of the atmospheric air being hitherto but little known, we have been satisfied to derive the name of its base from its known quality of killing such animals as are forced to breathe it, giving it the name of azot, from the Greek privitive particle α and ξωη, vita; hence the name of the noxious part of atmospheric air is azotic gas.»
- ^ Chaptal, J. A. and Nicholson, William trans. (1800) Elements of Chemistry, 3rd ed. London, England: C.C. and J. Robinson, vol. 1. pp. xxxv–xxxvi: «In order to correct the Nomenclature on this head [i.e., in this regard], nothing more is necessary than to substitute to [i.e., for] this word a denomination which is derived from the general system made use of; and I have presumed to propose that of Nitrogene Gas. In the first place, it is deduced from the characteristic and exclusive property of this gas, which forms the radical of the nitric acid. By this means we shall preserve to the combinations [i.e., compounds] of this substance the received [i.e., prevailing] denominations, such as those of the Nitric Acid, Nitrates, Nitrites, &c.»
- ^ nitrogen Archived 2017-07-02 at the Wayback Machine. Etymonline.com. Retrieved 2011-10-26.
- ^ Strutt, R. J. (1911) «Bakerian Lecture. A chemically active modification of nitrogen, produced by the electric discharge,» Archived 2016-12-20 at the Wayback Machine Proceedings of the Royal Society A, 85 (577): 219–29.
- ^ Lord Rayleigh’s Active Nitrogen Archived 2012-11-01 at the Wayback Machine. Lateralscience.co.uk. Retrieved 2011-10-26.
- ^ Erisman, Jan Willem; Sutton, Mark A.; Galloway, James; Klimont, Zbigniew; Winiwarter, Wilfried (2008). «How a century of ammonia synthesis changed the world». Nature Geoscience. 1 (10): 636. Bibcode:2008NatGe…1..636E. doi:10.1038/ngeo325. S2CID 94880859.
- ^ GB 190200698, Ostwald, Wilhelm, «Improvements in the Manufacture of Nitric Acid and Nitrogen Oxides», published 1902-03-20
- ^ GB 190208300, Ostwald, Wilhelm, «Improvements in and relating to the Manufacture of Nitric Acid and Oxides of Nitrogen», published 1903-02-26
- ^ a b c d e Greenwood and Earnshaw, pp. 411–12
- ^ Greenwood and Earnshaw, p. 550
- ^ Kaupp, Martin (1 December 2006). «The role of radial nodes of atomic orbitals for chemical bonding and the periodic table». Journal of Computational Chemistry. 28 (1): 320–25. doi:10.1002/jcc.20522. PMID 17143872. S2CID 12677737.
- ^ a b c d e f g h i j Greenwood and Earnshaw, pp. 412–16
- ^ Miller, T. S.; Belen, A.; Suter, T. M.; Sella, A.; Corà, A.; McMillan, P. F. (2017). «Carbon nitrides: synthesis and characterization of a new class of functional materials». Physical Chemistry Chemical Physics. 19 (24): 15613–15638. Bibcode:2017PCCP…1915613M. doi:10.1039/C7CP02711G. PMID 28594419.
- ^ House, J. E.; House, K. A. (2016). Descriptive Inorganic Chemistry. Amsterdam: Elsevier. p. 198. ISBN 978-0-12-804697-5.
- ^ Roy, A. K.; Burns, G. T.; Grigora, S.; Lie, G. C. (1994). «Poly(alkyl/aryloxothiazenes), [N=S(O)R]n : New direction in inorganic polymers». In Wisian-Neilson, P.; Alcock, H. R.; Wynne, K. J. (eds.). Inorganic and organometallic polymers II: advanced materials and intermediates. American Chemical Society. pp. 344–357. doi:10.1021/bk-1994-0572.ch026.
- ^ Holman, Jack P. (2002). Heat transfer (9th ed.). New York, NY: McGraw-Hill Companies, Inc. pp. 600–606. ISBN 9780072406559. OCLC 46959719.
- ^ Incropera 1 Dewitt 2 Bergman 3 Lavigne 4, Frank P. 1 David P. 2 Theodore L. 3 Adrienne S. 4 (2007). Fundamentals of heat and mass transfer (6th ed.). Hoboken, NJ: John Wiley and Sons, Inc. pp. 941–950. ISBN 9780471457282. OCLC 62532755.
- ^ Bethe, H. A. (1939). «Energy Production in Stars». Physical Review. 55 (5): 434–56. Bibcode:1939PhRv…55..434B. doi:10.1103/PhysRev.55.434. PMID 17835673.
- ^ CIAAW (2003). «Atomic Weight of Nitrogen». ciaaw.org. CIAAW. Archived from the original on 14 October 2016. Retrieved 13 October 2016.
- ^ Flanagan, Lawrence B.; Ehleringer, James R.; Pataki, Diane E. (15 December 2004). Stable Isotopes and Biosphere – Atmosphere Interactions: Processes and Biological Controls. pp. 74–75. ISBN 978-0-08-052528-0. Archived from the original on 5 February 2016. Retrieved 20 December 2015.
- ^ Greenwood and Earnshaw, p. 408
- ^ «Evaluated Nuclear Data File (ENDF) Retrieval & Plotting». National Nuclear Data Center. Archived from the original on 2020-08-09. Retrieved 2016-11-23.
- ^ Arthur G Palmer (2007). Protein NMR Spectroscopy. Elsevier Academic Press. ISBN 978-0-12-164491-8.
- ^ Katzenberg, M. A. (2008). «Chapter 13: Stable Isotope Analysis: A Tool for Studying Past Diet, Demography, and Life History». Biological Anthropology of the Human Skeleton (2nd ed.). ISBN 978-0-471-79372-4.
- ^ a b c Audi, Georges; Bersillon, Olivier; Blachot, Jean; Wapstra, Aaldert Hendrik (2003), «The NUBASE evaluation of nuclear and decay properties», Nuclear Physics A, 729: 3–128, Bibcode:2003NuPhA.729….3A, doi:10.1016/j.nuclphysa.2003.11.001
- ^ Carlson, Neil (January 22, 2012). Physiology of Behavior. Methods and Strategies of Research. Vol. 11th edition. Pearson. p. 151. ISBN 978-0-205-23939-9.
- ^ a b c Neeb, Karl Heinz (1997). The Radiochemistry of Nuclear Power Plants with Light Water Reactors. Berlin-New York: Walter de Gruyter. p. 227. ISBN 978-3-11-013242-7. Archived from the original on 2016-02-05. Retrieved 2015-12-20.
- ^ «Universal Industrial Gases, Inc…Nitrogen N2 Properties, Uses, Applications — Gas and Liquid».
- ^ Lewars, Errol G. (2008). Modeling Marvels: Computational Anticipation of Novel molecules. Springer Science+Business Media. pp. 141–63. doi:10.1007/978-1-4020-6973-4. ISBN 978-1-4020-6972-7.
- ^ «Polymeric nitrogen synthesized». physorg.com. 5 August 2004. Archived from the original on 2012-01-24. Retrieved 2009-06-22.
- ^ Gray, Theodore (2009). The Elements: A Visual Exploration of Every Known Atom in the Universe. New York: Black Dog & Leventhal Publishers. ISBN 978-1-57912-814-2.
- ^ Schuch, A. F.; Mills, R. L. (1970). «Crystal Structures of the Three Modifications of Nitrogen 14 and Nitrogen 15 at High Pressure». The Journal of Chemical Physics. 52 (12): 6000–08. Bibcode:1970JChPh..52.6000S. doi:10.1063/1.1672899.
- ^ Iancu, C. V.; Wright, E. R.; Heymann, J. B.; Jensen, G. J. (2006). «A comparison of liquid nitrogen and liquid helium as cryogens for electron cryotomography». Journal of Structural Biology. 153 (3): 231–40. doi:10.1016/j.jsb.2005.12.004. PMID 16427786.
- ^ «Flowing nitrogen ice glaciers seen on surface of Pluto after New Horizons flyby». ABC News. 25 July 2015. Archived from the original on 29 September 2015. Retrieved 6 October 2015.
- ^ McKinnon, William B.; Kirk, Randolph L. (2014). «Triton». In Spohn, Tilman; Breuer, Doris; Johnson, Torrence (eds.). Encyclopedia of the Solar System (3rd ed.). Amsterdam; Boston: Elsevier. pp. 861–82. ISBN 978-0-12-416034-7. Archived from the original on 2016-09-03. Retrieved 2016-04-30.
- ^ «Neptune: Moons: Triton». NASA. Archived from the original on October 15, 2011. Retrieved September 21, 2007.
- ^ Fryzuk, M. D. & Johnson, S. A. (2000). «The continuing story of dinitrogen activation». Coordination Chemistry Reviews. 200–202: 379. doi:10.1016/S0010-8545(00)00264-2.
- ^ Schrock, R. R. (2005). «Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center». Acc. Chem. Res. 38 (12): 955–62. doi:10.1021/ar0501121. PMC 2551323. PMID 16359167.
- ^ a b c d e Greenwood and Earnshaw, pp. 417–20
- ^ Greenwood and Earnshaw, pp. 434–38
- ^ Greenwood and Earnshaw, pp. 420–26
- ^ a b c d Greenwood and Earnshaw, pp. 426–33
- ^ Vieira, R.; C. Pham-Huu; N. Keller; M. J. Ledoux (2002). «New carbon nanofiber/graphite felt composite for use as a catalyst for hydrazine catalytic decomposition». Chemical Communications (9): 954–55. doi:10.1039/b202032g. PMID 12123065.
- ^ a b c d Greenwood and Earnshaw, pp. 438–42
- ^ Bowden, F. P. (1958). «Initiation of Explosion by Neutrons, α-Particles, and Fission Products». Proceedings of the Royal Society of London A. 246 (1245): 216–19. Bibcode:1958RSPSA.246..216B. doi:10.1098/rspa.1958.0123. S2CID 137728239.
- ^ Ford, L. A.; Grundmeier, E. W. (1993). Chemical Magic. Dover. p. 76. ISBN 978-0-486-67628-9.
- ^ Frierson, W. J.; Kronrad, J.; Browne, A. W. (1943). «Chlorine Azide, ClN3. I». Journal of the American Chemical Society. 65 (9): 1696–1698. doi:10.1021/ja01249a012.
- ^ Lyhs, Benjamin; Bläser, Dieter; Wölper, Christoph; Schulz, Stephan; Jansen, Georg (20 February 2012). «Solid-State Structure of Bromine Azide» (PDF). Angewandte Chemie International Edition. 51 (8): 1970–1974. doi:10.1002/anie.201108092. PMID 22250068. Archived (PDF) from the original on 25 August 2021. Retrieved 25 August 2021.
- ^ a b c d e f Greenwood and Earnshaw, pp. 443–58
- ^ Rahm, Martin; Dvinskikh, Sergey V.; Furó, István; Brinck, Tore (23 December 2010). «Experimental Detection of Trinitramide, N(NO2)3«. Angewandte Chemie International Edition. 50 (5): 1145–48. doi:10.1002/anie.201007047. PMID 21268214. S2CID 32952729.
- ^ Hou, Y. C.; Janczuk, A.; Wang, P. G. (1999). «Current trends in the development of nitric oxide donors». Current Pharmaceutical Design. 5 (6): 417–41. PMID 10390607.
- ^ Talawar, M. B.; et al. (2005). «Establishment of Process Technology for the Manufacture of Dinitrogen Pentoxide and its Utility for the Synthesis of Most Powerful Explosive of Today – CL-20». Journal of Hazardous Materials. 124 (1–3): 153–64. doi:10.1016/j.jhazmat.2005.04.021. PMID 15979786.
- ^ a b c d e f g h i Greenwood and Earnshaw, pp. 459–72
- ^ a b March, Jerry (1985), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (3rd ed.), New York: Wiley, ISBN 0-471-85472-7
- ^ Rédei, George P (2008). «Kjeldahl Method». Encyclopedia of Genetics, Genomics, Proteomics and Informatics. p. 1063. doi:10.1007/978-1-4020-6754-9_9066. ISBN 978-1-4020-6753-2.
- ^ Depending on the average thickness which is somewhere between 10 and 30 km, the mass of the earth’s crust is between 15×1018 and 45×1018 tonnes.
- ^ a b Greenwood and Earnshaw, pp. 407–09
- ^ Nielsen, M. K.; Jørgensen, B. M. (Jun 2004). «Quantitative relationship between trimethylamine oxide aldolase activity and formaldehyde accumulation in white muscle from gadiform fish during frozen storage». Journal of Agricultural and Food Chemistry. 52 (12): 3814–22. doi:10.1021/jf035169l. PMID 15186102.
- ^ Knox, G. A. (2007). Biology of the Southern Ocean. CRC Press. p. 392. ISBN 978-0-8493-3394-1. Archived from the original on 2021-10-01. Retrieved 2020-08-24.
- ^ Vickerstaff Joneja; Janice M. (2004). Digestion, diet, and disease: irritable bowel syndrome and gastrointestinal function. Rutgers University Press. p. 121. ISBN 978-0-8135-3387-2. Archived from the original on 2021-10-01. Retrieved 2020-08-24.
- ^ Froehlich, Peter (May 2013). «A Sustainable Approach to the Supply of Nitrogen». www.parker.com. Parker Hannifin Corporation. Archived from the original on 16 March 2016. Retrieved 24 November 2016.
- ^ Reich, Murray; Kapenekas, Harry (1957). «Nitrogen Purfication. Pilot Plant Removal of Oxygen». Industrial & Engineering Chemistry. 49 (5): 869–73. doi:10.1021/ie50569a032.
- ^ a b c d e Greenwood and Earnshaw, pp. 409–11
- ^ a b Bartlett, J. K. (1967). «Analysis for nitrite by evolution of nitrogen: A general chemistry laboratory experiment». Journal of Chemical Education. 44 (8): 475. Bibcode:1967JChEd..44..475B. doi:10.1021/ed044p475.
- ^ Eremets, M. I.; Popov, M. Y.; Trojan, I. A.; Denisov, V. N.; Boehler, R.; Hemley, R. J. (2004). «Polymerization of nitrogen in sodium azide». The Journal of Chemical Physics. 120 (22): 10618–23. Bibcode:2004JChPh.12010618E. doi:10.1063/1.1718250. PMID 15268087.
- ^ Ministers, Nordic Council of (2002). Food Additives in Europe 2000. p. 591. ISBN 978-92-893-0829-8. Archived from the original on 2016-02-05. Retrieved 2015-12-20.
- ^ Harding, Charlie, ed. (2002). Elements of the p Block. Cambridge: Royal Society of Chemistry. ISBN 978-0-85404-690-4. Archived from the original on 2021-10-01. Retrieved 2020-08-24.
- ^ Gavriliuk, V. G.; Berns, Hans (1999). High nitrogen steels: structure, properties, manufacture, applications. Springer. ISBN 978-3-540-66411-6. Archived from the original on 2021-10-01. Retrieved 2020-08-24.
- ^ Meka, S. R.; Chauhan, A.; Steiner, T.; Bischoff, E.; Ghosh, P. K.; Mittemeijer, E. J. (2015). «Generating duplex microstructures by nitriding; nitriding of iron based Fe–Mn alloy». Materials Science and Technology. 32 (9): 1743284715Y.000. doi:10.1179/1743284715Y.0000000098.
- ^ «Why don’t they use normal air in race car tires?». Howstuffworks. 2001-03-16. Archived from the original on 2011-07-12. Retrieved 2006-07-22.
- ^ Kemmochi, Y; Tsutsumi, K.; Arikawa, A.; Nakazawa, H. (2002). «Centrifugal concentrator for the substitution of nitrogen blow-down micro-concentration in dioxin/polychlorinated biphenyl sample preparation». Journal of Chromatography A. 943 (2): 295–97. doi:10.1016/S0021-9673(01)01466-2. PMID 11833649.
- ^ Baxter, E. Denise; Hughes, Paul S. (2001). Beer: Quality, Safety and Nutritional Aspects. Royal Society of Chemistry. p. 22. ISBN 978-0-85404-588-4. Archived from the original on 2020-03-21. Retrieved 2015-06-20.
- ^ «How does the widget in a beer can work?». Howstuffworks. 2000-08-16. Archived from the original on 2007-11-02. Retrieved 2008-07-30.
- ^ Denny, Mark (1 November 2009). Froth!: The Science of Beer. p. 131. ISBN 978-0-8018-9569-2. Archived from the original on 5 February 2016. Retrieved 20 December 2015.
- ^ Kennett, Andrew J. (2008). Design of a pneumatically assisted shifting system for Formula SAE® racing applications (Thesis). Dept. of Mechanical Engineering, Massachusetts Institute of Technology. hdl:1721.1/45820.
- ^ Betterton, E. A. (2003). «Environmental Fate of Sodium Azide Derived from Automobile Airbags». Critical Reviews in Environmental Science and Technology. 33 (4): 423–58. doi:10.1080/10643380390245002. S2CID 96404307.
- ^ Sanburn, Josh (2015-04-10). «The Dawn of a New Form of Capital Punishment». Time. Archived from the original on 2015-04-11. Retrieved 2015-04-11.
- ^ Sexton, Mike (18 December 2012). «Euthanasia campaigner under scrutiny». ABC. Archived from the original on 7 July 2013. Retrieved 6 May 2013.
- ^ Berman, Mark (April 17, 2015). «Oklahoma says it will now use nitrogen gas as its backup method of execution». The Washington Post. Archived from the original on June 23, 2019. Retrieved June 22, 2019.
- ^ «Oklahoma Attorney general says state will resume executions». New York Post. Archived from the original on March 9, 2021. Retrieved March 22, 2020.
- ^ Kaganer, M. G.; Kozheurov, V. & Levina, Zh. L. (1967). «Vessels for the storage and transport of liquid oxygen and nitrogen». Chemical and Petroleum Engineering. 3 (12): 918–22. doi:10.1007/BF01136404. S2CID 96762552.
- ^ Ahmed I; Agarwal S; Ilchyshyn A; Charles-Holmes S; Berth-Jones J (May 2001). «Liquid nitrogen cryotherapy of common warts: cryo-spray vs. cotton wool bud». Br. J. Dermatol. 144 (5): 1006–09. doi:10.1046/j.1365-2133.2001.04190.x. PMID 11359389. S2CID 221325640.
- ^ «Biology Safety – Cryogenic materials. The risks posed by them». University of Bath. Archived from the original on February 6, 2007. Retrieved 2007-01-03.
- ^ «Space Shuttle Columbia Fast Facts». CNN. September 30, 2013. Archived from the original on February 2, 2016. Retrieved January 20, 2016.
- ^ Fowler, B.; Ackles, K. N.; Porlier, G. (1985). «Effects of inert gas narcosis on behavior – a critical review». Undersea Biomed. Res. 12 (4): 369–402. PMID 4082343. Archived from the original on 2010-12-25. Retrieved 2008-09-21.
{{cite journal}}: CS1 maint: unfit URL (link) - ^ Rogers, W. H.; Moeller, G. (1989). «Effect of brief, repeated hyperbaric exposures on susceptibility to nitrogen narcosis». Undersea Biomed. Res. 16 (3): 227–32. OCLC 2068005. PMID 2741255. Archived from the original on 2009-09-01. Retrieved 2008-09-21.
{{cite journal}}: CS1 maint: unfit URL (link) - ^ Acott, C. (1999). «A brief history of diving and decompression illness». South Pacific Underwater Medicine Society Journal. 29 (2). OCLC 16986801. Archived from the original on 2011-09-05. Retrieved 2008-09-21.
{{cite journal}}: CS1 maint: unfit URL (link) - ^ Kindwall, E. P.; Baz, A.; Lightfoot, E. N.; Lanphier, E. H.; Seireg, A. (1975). «Nitrogen elimination in man during decompression». Undersea Biomed. Res. 2 (4): 285–97. OCLC 2068005. PMID 1226586. Archived from the original on 2011-07-27. Retrieved 2008-09-21.
{{cite journal}}: CS1 maint: unfit URL (link) - ^ US Navy Diving Manual, 6th revision. United States: US Naval Sea Systems Command. 2006. Archived from the original on 2008-05-02. Retrieved 2008-04-24.
- ^ Walker, Jearl. «Boiling and the Leidenfrost Effect» (PDF). Fundamentals of Physics: 1–4. Archived (PDF) from the original on 13 December 2019. Retrieved 11 October 2014.
- ^ Liquid nitrogen cocktail leaves teen in hospital Archived 2017-04-12 at the Wayback Machine, BBC News, October 8, 2012.
- ^ Mattox, Brent S. «Investigative Report on Chemistry 301A Cylinder Explosion» (PDF). Texas A&M University. Archived from the original (reprint) on 2014-04-30.
- ^ British Compressed Gases Association (2000) BCGA Code of Practice CP30. The Safe Use of Liquid nitrogen Dewars up to 50 litres. Archived 2007-07-18 at the Wayback Machine ISSN 0260-4809.
- ^ Confined Space Entry – Worker and Would-be Rescuer Asphyxiated Archived 2015-09-22 at the Wayback Machine, Valero Refinery Asphyxiation Incident Case Study.
- ^ Inquiry after man dies in chemical leak Archived 2017-01-07 at the Wayback Machine, BBC News, October 25, 1999.
- ^ Liquid Nitrogen – Code of practice for handling. United Kingdom: Birkbeck, University of London. 2007. Archived from the original on 2018-06-12. Retrieved 2012-02-08.
- ^ Levey, Christopher G. «Liquid Nitrogen Safety». Thayer School of Engineering at Dartmouth. Archived from the original on 2016-03-05. Retrieved 2016-11-23.
- ^ National Institutes of Health. Protocol for Use and Maintenance of Oxygen Monitoring Devices. February 2014, at 1:35 UTC. Available at: https://www.ors.od.nih.gov/sr/dohs/documents/protocoloxygenmonitoring.pdf Archived 2020-12-05 at the Wayback Machine. Accessed June 23, 2020
Bibliography
- Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 978-0-08-037941-8.
External links
- Etymology of Nitrogen
- Nitrogen at The Periodic Table of Videos (University of Nottingham)
- Nitrogen podcast from the Royal Society of Chemistry’s Chemistry World
| Liquid nitrogen (N2 at < −196 °C) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Nitrogen | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allotropes | see § Allotropes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Appearance | colorless gas, liquid or solid | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Standard atomic weight Ar°(N) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Nitrogen in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Group | group 15 (pnictogens) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Period | period 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Block | p-block | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electron configuration | [He] 2s2 2p3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Physical properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phase at STP | gas | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Melting point | (N2) 63.23[2] K (−209.86[2] °C, −345.75[2] °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Boiling point | (N2) 77.355 K (−195.795 °C, −320.431 °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density (at STP) | 1.2506 g/L[3] at 0 °C, 1013 mbar | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| when liquid (at b.p.) | 0.808 g/cm3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Triple point | 63.151 K, 12.52 kPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Critical point | 126.21 K, 3.39 MPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of fusion | (N2) 0.72 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of vaporisation | (N2) 5.57 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar heat capacity | (N2) 29.124 J/(mol·K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Vapour pressure
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxidation states | −3, −2, −1, 0,[4] +1, +2, +3, +4, +5 (a strongly acidic oxide) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 3.04 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ionisation energies |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Covalent radius | 71±1 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Van der Waals radius | 155 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Spectral lines of nitrogen | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Natural occurrence | primordial | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure | hexagonal
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Speed of sound | 353 m/s (gas, at 27 °C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal conductivity | 25.83×10−3 W/(m⋅K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Magnetic ordering | diamagnetic | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAS Number | 17778-88-0 7727-37-9 (N2) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| History | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Discovery | Daniel Rutherford (1772) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Named by | Jean-Antoine Chaptal (1790) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Main isotopes of nitrogen
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| references |
Nitrogen is the chemical element with the symbol N and atomic number 7. Nitrogen is a nonmetal and the lightest member of group 15 of the periodic table, often called the pnictogens. It is a common element in the universe, estimated at seventh in total abundance in the Milky Way and the Solar System. At standard temperature and pressure, two atoms of the element bond to form N2, a colorless and odorless diatomic gas. N2 forms about 78% of Earth’s atmosphere, making it the most abundant uncombined element. Nitrogen occurs in all organisms, primarily in amino acids (and thus proteins), in the nucleic acids (DNA and RNA) and in the energy transfer molecule adenosine triphosphate. The human body contains about 3% nitrogen by mass, the fourth most abundant element in the body after oxygen, carbon, and hydrogen. The nitrogen cycle describes the movement of the element from the air, into the biosphere and organic compounds, then back into the atmosphere.
Many industrially important compounds, such as ammonia, nitric acid, organic nitrates (propellants and explosives), and cyanides, contain nitrogen. The extremely strong triple bond in elemental nitrogen (N≡N), the second strongest bond in any diatomic molecule after carbon monoxide (CO),[5] dominates nitrogen chemistry. This causes difficulty for both organisms and industry in converting N2 into useful compounds, but at the same time it means that burning, exploding, or decomposing nitrogen compounds to form nitrogen gas releases large amounts of often useful energy. Synthetically produced ammonia and nitrates are key industrial fertilisers, and fertiliser nitrates are key pollutants in the eutrophication of water systems.
It was first discovered and isolated by Scottish physician Daniel Rutherford in 1772. Although Carl Wilhelm Scheele and Henry Cavendish had independently done so at about the same time, Rutherford is generally accorded the credit because his work was published first. The name nitrogène was suggested by French chemist Jean-Antoine-Claude Chaptal in 1790 when it was found that nitrogen was present in nitric acid and nitrates. Antoine Lavoisier suggested instead the name azote, from the Ancient Greek: ἀζωτικός «no life», as it is an asphyxiant gas; this name is used in several languages, including French, Italian, Russian, Romanian, Portuguese and Turkish, and appears in the English names of some nitrogen compounds such as hydrazine, azides and azo compounds.
Apart from its use in fertilisers and energy stores, nitrogen is a constituent of organic compounds as diverse as Kevlar used in high-strength fabric and cyanoacrylate used in superglue. Nitrogen is a constituent of every major pharmacological drug class, including antibiotics. Many drugs are mimics or prodrugs of natural nitrogen-containing signal molecules: for example, the organic nitrates nitroglycerin and nitroprusside control blood pressure by metabolizing into nitric oxide. Many notable nitrogen-containing drugs, such as the natural caffeine and morphine or the synthetic amphetamines, act on receptors of animal neurotransmitters.
History
Nitrogen compounds have a very long history, ammonium chloride having been known to Herodotus. They were well-known by the Middle Ages. Alchemists knew nitric acid as aqua fortis (strong water), as well as other nitrogen compounds such as ammonium salts and nitrate salts. The mixture of nitric and hydrochloric acids was known as aqua regia (royal water), celebrated for its ability to dissolve gold, the king of metals.[6]
The discovery of nitrogen is attributed to the Scottish physician Daniel Rutherford in 1772, who called it noxious air.[7][8] Though he did not recognise it as an entirely different chemical substance, he clearly distinguished it from Joseph Black’s «fixed air», or carbon dioxide.[9] The fact that there was a component of air that does not support combustion was clear to Rutherford, although he was not aware that it was an element. Nitrogen was also studied at about the same time by Carl Wilhelm Scheele,[10] Henry Cavendish,[11] and Joseph Priestley,[12] who referred to it as burnt air or phlogisticated air. French chemist Antoine Lavoisier referred to nitrogen gas as «mephitic air» or azote, from the Greek word άζωτικός (azotikos), «no life», due to it being asphyxiant.[13][14] In an atmosphere of pure nitrogen, animals died and flames were extinguished. Though Lavoisier’s name was not accepted in English since it was pointed out that all gases but oxygen are either asphyxiant or outright toxic, it is used in many languages (French, Italian, Portuguese, Polish, Russian, Albanian, Turkish, etc.; the German Stickstoff similarly refers to the same characteristic, viz. ersticken «to choke or suffocate») and still remains in English in the common names of many nitrogen compounds, such as hydrazine and compounds of the azide ion. Finally, it led to the name «pnictogens» for the group headed by nitrogen, from the Greek πνίγειν «to choke».[6]
The English word nitrogen (1794) entered the language from the French nitrogène, coined in 1790 by French chemist Jean-Antoine Chaptal (1756–1832),[15] from the French nitre (potassium nitrate, also called saltpeter) and the French suffix -gène, «producing», from the Greek -γενής (-genes, «begotten»). Chaptal’s meaning was that nitrogen is the essential part of nitric acid, which in turn was produced from nitre. In earlier times, niter had been confused with Egyptian «natron» (sodium carbonate) – called νίτρον (nitron) in Greek – which, despite the name, contained no nitrate.[16]
The earliest military, industrial, and agricultural applications of nitrogen compounds used saltpeter (sodium nitrate or potassium nitrate), most notably in gunpowder, and later as fertiliser. In 1910, Lord Rayleigh discovered that an electrical discharge in nitrogen gas produced «active nitrogen», a monatomic allotrope of nitrogen.[17] The «whirling cloud of brilliant yellow light» produced by his apparatus reacted with mercury to produce explosive mercury nitride.[18]
For a long time, sources of nitrogen compounds were limited. Natural sources originated either from biology or deposits of nitrates produced by atmospheric reactions. Nitrogen fixation by industrial processes like the Frank–Caro process (1895–1899) and Haber–Bosch process (1908–1913) eased this shortage of nitrogen compounds, to the extent that half of global food production (see Applications) now relies on synthetic nitrogen fertilisers.[19] At the same time, use of the Ostwald process (1902) to produce nitrates from industrial nitrogen fixation allowed the large-scale industrial production of nitrates as feedstock in the manufacture of explosives in the World Wars of the 20th century.[20][21]
Properties
Atomic
The shapes of the five orbitals occupied in nitrogen. The two colours show the phase or sign of the wave function in each region. From left to right: 1s, 2s (cutaway to show internal structure), 2px, 2py, 2pz.
A nitrogen atom has seven electrons. In the ground state, they are arranged in the electron configuration 1s2
2s2
2p1
x2p1
y2p1
z. It, therefore, has five valence electrons in the 2s and 2p orbitals, three of which (the p-electrons) are unpaired. It has one of the highest electronegativities among the elements (3.04 on the Pauling scale), exceeded only by chlorine (3.16), oxygen (3.44), and fluorine (3.98). (The light noble gases, helium, neon, and argon, would presumably also be more electronegative, and in fact are on the Allen scale.)[22] Following periodic trends, its single-bond covalent radius of 71 pm is smaller than those of boron (84 pm) and carbon (76 pm), while it is larger than those of oxygen (66 pm) and fluorine (57 pm). The nitride anion, N3−, is much larger at 146 pm, similar to that of the oxide (O2−: 140 pm) and fluoride (F−: 133 pm) anions.[22] The first three ionisation energies of nitrogen are 1.402, 2.856, and 4.577 MJ·mol−1, and the sum of the fourth and fifth is 16.920 MJ·mol−1. Due to these very high figures, nitrogen has no simple cationic chemistry.[23]
The lack of radial nodes in the 2p subshell is directly responsible for many of the anomalous properties of the first row of the p-block, especially in nitrogen, oxygen, and fluorine. The 2p subshell is very small and has a very similar radius to the 2s shell, facilitating orbital hybridisation. It also results in very large electrostatic forces of attraction between the nucleus and the valence electrons in the 2s and 2p shells, resulting in very high electronegativities. Hypervalency is almost unknown in the 2p elements for the same reason, because the high electronegativity makes it difficult for a small nitrogen atom to be a central atom in an electron-rich three-center four-electron bond since it would tend to attract the electrons strongly to itself. Thus, despite nitrogen’s position at the head of group 15 in the periodic table, its chemistry shows huge differences from that of its heavier congeners phosphorus, arsenic, antimony, and bismuth.[24]
Nitrogen may be usefully compared to its horizontal neighbours’ carbon and oxygen as well as its vertical neighbours in the pnictogen column, phosphorus, arsenic, antimony, and bismuth. Although each period 2 element from lithium to oxygen shows some similarities to the period 3 element in the next group (from magnesium to chlorine; these are known as diagonal relationships), their degree drops off abruptly past the boron–silicon pair. The similarities of nitrogen to sulfur are mostly limited to sulfur nitride ring compounds when both elements are the only ones present.[25]
Nitrogen does not share the proclivity of carbon for catenation. Like carbon, nitrogen tends to form ionic or metallic compounds with metals. Nitrogen forms an extensive series of nitrides with carbon, including those with chain-, graphitic-, and fullerenic-like structures.[26]
It resembles oxygen with its high electronegativity and concomitant capability for hydrogen bonding and the ability to form coordination complexes by donating its lone pairs of electrons. There are some parallels between the chemistry of ammonia NH3 and water H2O. For example, the capacity of both compounds to be protonated to give NH4+ and H3O+ or deprotonated to give NH2− and OH−, with all of these able to be isolated in solid compounds.[27]
Nitrogen shares with both its horizontal neighbours a preference for forming multiple bonds, typically with carbon, oxygen, or other nitrogen atoms, through pπ–pπ interactions.[25] Thus, for example, nitrogen occurs as diatomic molecules and therefore has very much lower melting (−210 °C) and boiling points (−196 °C) than the rest of its group, as the N2 molecules are only held together by weak van der Waals interactions and there are very few electrons available to create significant instantaneous dipoles. This is not possible for its vertical neighbours; thus, the nitrogen oxides, nitrites, nitrates, nitro-, nitroso-, azo-, and diazo-compounds, azides, cyanates, thiocyanates, and imino-derivatives find no echo with phosphorus, arsenic, antimony, or bismuth. By the same token, however, the complexity of the phosphorus oxoacids finds no echo with nitrogen.[25] Setting aside their differences, nitrogen and phosphorus form an extensive series of compounds with one another; these have chain, ring, and cage structures.[28]
Table of thermal and physical properties of nitrogen (N2) at atmospheric pressure:[29][30]
| Temperature (K) | Density (kg/m^3) | Specific heat (kJ/kg °C) | Dynamic viscosity (kg/m s) | Kinematic viscosity (m^2/s) | Thermal conductivity (W/m °C) | Thermal diffusivity (m^2/s) | Prandtl Number |
| 100 | 3.4388 | 1.07 | 6.88E-06 | 2.00E-06 | 0.00958 | 2.60E-06 | 0.768 |
| 150 | 2.2594 | 1.05 | 1.01E-05 | 4.45E-06 | 0.0139 | 5.86E-06 | 0.759 |
| 200 | 1.7108 | 1.0429 | 1.29E-05 | 7.57E-06 | 0.01824 | 1.02E-05 | 0.747 |
| 300 | 1.1421 | 1.0408 | 1.78E-05 | 1.56E-05 | 0.0262 | 2.20E-05 | 0.713 |
| 400 | 0.8538 | 1.0459 | 2.20E-05 | 2.57E-05 | 0.03335 | 3.73E-05 | 0.691 |
| 500 | 0.6824 | 1.0555 | 2.57E-05 | 3.77E-05 | 0.03984 | 5.53E-05 | 0.684 |
| 600 | 0.5687 | 1.0756 | 2.91E-05 | 5.12E-05 | 0.0458 | 7.49E-05 | 0.686 |
| 700 | 0.4934 | 1.0969 | 3.21E-05 | 6.67E-05 | 0.05123 | 9.47E-05 | 0.691 |
| 800 | 0.4277 | 1.1225 | 3.48E-05 | 8.15E-05 | 0.05609 | 1.17E-04 | 0.7 |
| 900 | 0.3796 | 1.1464 | 3.75E-05 | 9.11E-05 | 0.0607 | 1.39E-04 | 0.711 |
| 1000 | 0.3412 | 1.1677 | 4.00E-05 | 1.19E-04 | 0.06475 | 1.63E-04 | 0.724 |
| 1100 | 0.3108 | 1.1857 | 4.23E-05 | 1.36E-04 | 0.0685 | 1.86E-04 | 0.736 |
| 1200 | 0.2851 | 1.2037 | 4.45E-05 | 1.56E-04 | 0.07184 | 2.09E-04 | 0.748 |
| 1300 | 0.2591 | 1.219 | 4.66E-05 | 1.80E-04 | 0.081 | 2.56E-04 | 0.701 |
Isotopes
Table of nuclides (Segrè chart) from carbon to fluorine (including nitrogen). Orange indicates proton emission (nuclides outside the proton drip line); pink for positron emission (inverse beta decay); black for stable nuclides; blue for electron emission (beta decay); and violet for neutron emission (nuclides outside the neutron drip line). Proton number increases going up the vertical axis and neutron number going to the right on the horizontal axis.
Nitrogen has two stable isotopes: 14N and 15N. The first is much more common, making up 99.634% of natural nitrogen, and the second (which is slightly heavier) makes up the remaining 0.366%. This leads to an atomic weight of around 14.007 u.[22] Both of these stable isotopes are produced in the CNO cycle in stars, but 14N is more common as its neutron capture is the rate-limiting step. 14N is one of the five stable odd–odd nuclides (a nuclide having an odd number of protons and neutrons); the other four are 2H, 6Li, 10B, and 180mTa.[31]
The relative abundance of 14N and 15N is practically constant in the atmosphere but can vary elsewhere, due to natural isotopic fractionation from biological redox reactions and the evaporation of natural ammonia or nitric acid.[32] Biologically mediated reactions (e.g., assimilation, nitrification, and denitrification) strongly control nitrogen dynamics in the soil. These reactions typically result in 15N enrichment of the substrate and depletion of the product.[33]
The heavy isotope 15N was first discovered by S. M. Naudé in 1929, and soon after heavy isotopes of the neighbouring elements oxygen and carbon were discovered.[34] It presents one of the lowest thermal neutron capture cross-sections of all isotopes.[35] It is frequently used in nuclear magnetic resonance (NMR) spectroscopy to determine the structures of nitrogen-containing molecules, due to its fractional nuclear spin of one-half, which offers advantages for NMR such as narrower line width. 14N, though also theoretically usable, has an integer nuclear spin of one and thus has a quadrupole moment that leads to wider and less useful spectra.[22] 15N NMR nevertheless has complications not encountered in the more common 1H and 13C NMR spectroscopy. The low natural abundance of 15N (0.36%) significantly reduces sensitivity, a problem which is only exacerbated by its low gyromagnetic ratio, (only 10.14% that of 1H). As a result, the signal-to-noise ratio for 1H is about 300 times as much as that for 15N at the same magnetic field strength.[36] This may be somewhat alleviated by isotopic enrichment of 15N by chemical exchange or fractional distillation. 15N-enriched compounds have the advantage that under standard conditions, they do not undergo chemical exchange of their nitrogen atoms with atmospheric nitrogen, unlike compounds with labelled hydrogen, carbon, and oxygen isotopes that must be kept away from the atmosphere.[22] The 15N:14N ratio is commonly used in stable isotope analysis in the fields of geochemistry, hydrology, paleoclimatology and paleoceanography, where it is called δ15N.[37]
Of the ten other isotopes produced synthetically, ranging from 12N to 23N, 13N has a half-life of ten minutes and the remaining isotopes have half-lives on the order of seconds (16N and 17N) or milliseconds. No other nitrogen isotopes are possible as they would fall outside the nuclear drip lines, leaking out a proton or neutron.[38] Given the half-life difference, 13N is the most important nitrogen radioisotope, being relatively long-lived enough to use in positron emission tomography (PET), although its half-life is still short and thus it must be produced at the venue of the PET, for example in a cyclotron via proton bombardment of 16O producing 13N and an alpha particle.[39]
The radioisotope 16N is the dominant radionuclide in the coolant of pressurised water reactors or boiling water reactors during normal operation. It is produced from 16O (in water) via an (n,p) reaction, in which the 16O atom captures a neutron and expels a proton. It has a short half-life of about 7.1 s,[38] but during its decay back to 16O produces high-energy gamma radiation (5 to 7 MeV).[38][40] Because of this, access to the primary coolant piping in a pressurised water reactor must be restricted during reactor power operation.[40] It is a sensitive and immediate indicator of leaks from the primary coolant system to the secondary steam cycle and is the primary means of detection for such leaks.[40]
Chemistry and compounds
Allotropes
Molecular orbital diagram of dinitrogen molecule, N2. There are five bonding orbitals and two antibonding orbitals (marked with an asterisk; orbitals involving the inner 1s electrons not shown), giving a total bond order of three.
Atomic nitrogen, also known as active nitrogen, is highly reactive, being a triradical with three unpaired electrons. Free nitrogen atoms easily react with most elements to form nitrides, and even when two free nitrogen atoms collide to produce an excited N2 molecule, they may release so much energy on collision with even such stable molecules as carbon dioxide and water to cause homolytic fission into radicals such as CO and O or OH and H. Atomic nitrogen is prepared by passing an electric discharge through nitrogen gas at 0.1–2 mmHg, which produces atomic nitrogen along with a peach-yellow emission that fades slowly as an afterglow for several minutes even after the discharge terminates.[25]
Given the great reactivity of atomic nitrogen, elemental nitrogen usually occurs as molecular N2, dinitrogen. This molecule is a colourless, odourless, and tasteless diamagnetic gas at standard conditions: it melts at −210 °C and boils at −196 °C.[25] Dinitrogen is mostly unreactive at room temperature, but it will nevertheless react with lithium metal and some transition metal complexes. This is due to its bonding, which is unique among the diatomic elements at standard conditions in that it has an N≡N triple bond. Triple bonds have short bond lengths (in this case, 109.76 pm) and high dissociation energies (in this case, 945.41 kJ/mol), and are thus very strong, explaining dinitrogen’s low level of chemical reactivity.[25][41]
Other nitrogen oligomers and polymers may be possible. If they could be synthesised, they may have potential applications as materials with a very high energy density, that could be used as powerful propellants or explosives.[42] Under extremely high pressures (1.1 million atm) and high temperatures (2000 K), as produced in a diamond anvil cell, nitrogen polymerises into the single-bonded cubic gauche crystal structure. This structure is similar to that of diamond, and both have extremely strong covalent bonds, resulting in its nickname «nitrogen diamond».[43]
At atmospheric pressure, molecular nitrogen condenses (liquefies) at 77 K (−195.79 °C) and freezes at 63 K (−210.01 °C)[44] into the beta hexagonal close-packed crystal allotropic form. Below 35.4 K (−237.6 °C) nitrogen assumes the cubic crystal allotropic form (called the alpha phase).[45] Liquid nitrogen, a colourless fluid resembling water in appearance, but with 80.8% of the density (the density of liquid nitrogen at its boiling point is 0.808 g/mL), is a common cryogen.[46] Solid nitrogen has many crystalline modifications. It forms a significant dynamic surface coverage on Pluto[47] and outer moons of the Solar System such as Triton.[48] Even at the low temperatures of solid nitrogen it is fairly volatile and can sublime to form an atmosphere, or condense back into nitrogen frost. It is very weak and flows in the form of glaciers and on Triton geysers of nitrogen gas come from the polar ice cap region.[49]
Dinitrogen complexes
The first example of a dinitrogen complex to be discovered was [Ru(NH3)5(N2)]2+ (see figure at right), and soon many other such complexes were discovered. These complexes, in which a nitrogen molecule donates at least one lone pair of electrons to a central metal cation, illustrate how N2 might bind to the metal(s) in nitrogenase and the catalyst for the Haber process: these processes involving dinitrogen activation are vitally important in biology and in the production of fertilisers.[50][51]
Dinitrogen is able to coordinate to metals in five different ways. The more well-characterised ways are the end-on M←N≡N (η1) and M←N≡N→M (μ, bis-η1), in which the lone pairs on the nitrogen atoms are donated to the metal cation. The less well-characterised ways involve dinitrogen donating electron pairs from the triple bond, either as a bridging ligand to two metal cations (μ, bis-η2) or to just one (η2). The fifth and unique method involves triple-coordination as a bridging ligand, donating all three electron pairs from the triple bond (μ3-N2). A few complexes feature multiple N2 ligands and some feature N2 bonded in multiple ways. Since N2 is isoelectronic with carbon monoxide (CO) and acetylene (C2H2), the bonding in dinitrogen complexes is closely allied to that in carbonyl compounds, although N2 is a weaker σ-donor and π-acceptor than CO. Theoretical studies show that σ donation is a more important factor allowing the formation of the M–N bond than π back-donation, which mostly only weakens the N–N bond, and end-on (η1) donation is more readily accomplished than side-on (η2) donation.[25]
Today, dinitrogen complexes are known for almost all the transition metals, accounting for several hundred compounds. They are normally prepared by three methods:[25]
- Replacing labile ligands such as H2O, H−, or CO directly by nitrogen: these are often reversible reactions that proceed at mild conditions.
- Reducing metal complexes in the presence of a suitable co-ligand in excess under nitrogen gas. A common choice includes replacing chloride ligands with dimethylphenylphosphine (PMe2Ph) to make up for the smaller number of nitrogen ligands attached to the original chlorine ligands.
- Converting a ligand with N–N bonds, such as hydrazine or azide, directly into a dinitrogen ligand.
Occasionally the N≡N bond may be formed directly within a metal complex, for example by directly reacting coordinated ammonia (NH3) with nitrous acid (HNO2), but this is not generally applicable. Most dinitrogen complexes have colours within the range white-yellow-orange-red-brown; a few exceptions are known, such as the blue [{Ti(η5-C5H5)2}2-(N2)].[25]
Nitrides, azides, and nitrido complexes
Nitrogen bonds to almost all the elements in the periodic table except the first three noble gases, helium, neon, and argon, and some of the very short-lived elements after bismuth, creating an immense variety of binary compounds with varying properties and applications in which pentazenium tetraazidoborate has the highest nitrogen content.[25] Many binary compounds are known: with the exception of the nitrogen hydrides, oxides, and fluorides, these are typically called nitrides. Many stoichiometric phases are usually present for most elements (e.g. MnN, Mn6N5, Mn3N2, Mn2N, Mn4N, and MnxN for 9.2 < x < 25.3). They may be classified as «salt-like» (mostly ionic), covalent, «diamond-like», and metallic (or interstitial), although this classification has limitations generally stemming from the continuity of bonding types instead of the discrete and separate types that it implies. They are normally prepared by directly reacting a metal with nitrogen or ammonia (sometimes after heating), or by thermal decomposition of metal amides:[52]
- 3 Ca + N2 → Ca3N2
- 3 Mg + 2 NH3 → Mg3N2 + 3 H2 (at 900 °C)
- 3 Zn(NH2)2 → Zn3N2 + 4 NH3
Many variants on these processes are possible. The most ionic of these nitrides are those of the alkali metals and alkaline earth metals, Li3N (Na, K, Rb, and Cs do not form stable nitrides for steric reasons) and M3N2 (M = Be, Mg, Ca, Sr, Ba). These can formally be thought of as salts of the N3− anion, although charge separation is not actually complete even for these highly electropositive elements. However, the alkali metal azides NaN3 and KN3, featuring the linear N−
3 anion, are well-known, as are Sr(N3)2 and Ba(N3)2. Azides of the B-subgroup metals (those in groups 11 through 16) are much less ionic, have more complicated structures, and detonate readily when shocked.[52]
Mesomeric structures of borazine, (–BH–NH–)3
Many covalent binary nitrides are known. Examples include cyanogen ((CN)2), triphosphorus pentanitride (P3N5), disulfur dinitride (S2N2), and tetrasulfur tetranitride (S4N4). The essentially covalent silicon nitride (Si3N4) and germanium nitride (Ge3N4) are also known: silicon nitride, in particular, would make a promising ceramic if not for the difficulty of working with and sintering it. In particular, the group 13 nitrides, most of which are promising semiconductors, are isoelectronic with graphite, diamond, and silicon carbide and have similar structures: their bonding changes from covalent to partially ionic to metallic as the group is descended. In particular, since the B–N unit is isoelectronic to C–C, and carbon is essentially intermediate in size between boron and nitrogen, much of organic chemistry finds an echo in boron–nitrogen chemistry, such as in borazine («inorganic benzene»). Nevertheless, the analogy is not exact due to the ease of nucleophilic attack at boron due to its deficiency in electrons, which is not possible in a wholly carbon-containing ring.[52]
The largest category of nitrides are the interstitial nitrides of formulae MN, M2N, and M4N (although variable composition is perfectly possible), where the small nitrogen atoms are positioned in the gaps in a metallic cubic or hexagonal close-packed lattice. They are opaque, very hard, and chemically inert, melting only at very high temperatures (generally over 2500 °C). They have a metallic lustre and conduct electricity as do metals. They hydrolyse only very slowly to give ammonia or nitrogen.[52]
The nitride anion (N3−) is the strongest π donor known among ligands (the second-strongest is O2−). Nitrido complexes are generally made by the thermal decomposition of azides or by deprotonating ammonia, and they usually involve a terminal {≡N}3− group. The linear azide anion (N−
3), being isoelectronic with nitrous oxide, carbon dioxide, and cyanate, forms many coordination complexes. Further catenation is rare, although N4−
4 (isoelectronic with carbonate and nitrate) is known.[52]
Hydrides
Standard reduction potentials for nitrogen-containing species. Top diagram shows potentials at pH 0; bottom diagram shows potentials at pH 14.[53]
Industrially, ammonia (NH3) is the most important compound of nitrogen and is prepared in larger amounts than any other compound because it contributes significantly to the nutritional needs of terrestrial organisms by serving as a precursor to food and fertilisers. It is a colourless alkaline gas with a characteristic pungent smell. The presence of hydrogen bonding has very significant effects on ammonia, conferring on it its high melting (−78 °C) and boiling (−33 °C) points. As a liquid, it is a very good solvent with a high heat of vaporisation (enabling it to be used in vacuum flasks), that also has a low viscosity and electrical conductivity and high dielectric constant, and is less dense than water. However, the hydrogen bonding in NH3 is weaker than that in H2O due to the lower electronegativity of nitrogen compared to oxygen and the presence of only one lone pair in NH3 rather than two in H2O. It is a weak base in aqueous solution (pKb 4.74); its conjugate acid is ammonium, NH+
4. It can also act as an extremely weak acid, losing a proton to produce the amide anion, NH−
2. It thus undergoes self-dissociation, similar to water, to produce ammonium and amide. Ammonia burns in air or oxygen, though not readily, to produce nitrogen gas; it burns in fluorine with a greenish-yellow flame to give nitrogen trifluoride. Reactions with the other nonmetals are very complex and tend to lead to a mixture of products. Ammonia reacts on heating with metals to give nitrides.[54]
Many other binary nitrogen hydrides are known, but the most important are hydrazine (N2H4) and hydrogen azide (HN3). Although it is not a nitrogen hydride, hydroxylamine (NH2OH) is similar in properties and structure to ammonia and hydrazine as well. Hydrazine is a fuming, colourless liquid that smells similar to ammonia. Its physical properties are very similar to those of water (melting point 2.0 °C, boiling point 113.5 °C, density 1.00 g/cm3). Despite it being an endothermic compound, it is kinetically stable. It burns quickly and completely in air very exothermically to give nitrogen and water vapour. It is a very useful and versatile reducing agent and is a weaker base than ammonia.[55] It is also commonly used as a rocket fuel.[56]
Hydrazine is generally made by reaction of ammonia with alkaline sodium hypochlorite in the presence of gelatin or glue:[55]
- NH3 + OCl− → NH2Cl + OH−
- NH2Cl + NH3 → N
2H+
5 + Cl− (slow) - N
2H+
5 + OH− → N2H4 + H2O (fast)
(The attacks by hydroxide and ammonia may be reversed, thus passing through the intermediate NHCl− instead.) The reason for adding gelatin is that it removes metal ions such as Cu2+ that catalyses the destruction of hydrazine by reaction with monochloramine (NH2Cl) to produce ammonium chloride and nitrogen.[55]
Hydrogen azide (HN3) was first produced in 1890 by the oxidation of aqueous hydrazine by nitrous acid. It is very explosive and even dilute solutions can be dangerous. It has a disagreeable and irritating smell and is a potentially lethal (but not cumulative) poison. It may be considered the conjugate acid of the azide anion, and is similarly analogous to the hydrohalic acids.[55]
Halides and oxohalides
All four simple nitrogen trihalides are known. A few mixed halides and hydrohalides are known, but are mostly unstable; examples include NClF2, NCl2F, NBrF2, NF2H, NFH2, NCl2H, and NClH2.[57]
Five nitrogen fluorides are known. Nitrogen trifluoride (NF3, first prepared in 1928) is a colourless and odourless gas that is thermodynamically stable, and most readily produced by the electrolysis of molten ammonium fluoride dissolved in anhydrous hydrogen fluoride. Like carbon tetrafluoride, it is not at all reactive and is stable in water or dilute aqueous acids or alkalis. Only when heated does it act as a fluorinating agent, and it reacts with copper, arsenic, antimony, and bismuth on contact at high temperatures to give tetrafluorohydrazine (N2F4). The cations NF+
4 and N
2F+
3 are also known (the latter from reacting tetrafluorohydrazine with strong fluoride-acceptors such as arsenic pentafluoride), as is ONF3, which has aroused interest due to the short N–O distance implying partial double bonding and the highly polar and long N–F bond. Tetrafluorohydrazine, unlike hydrazine itself, can dissociate at room temperature and above to give the radical NF2•. Fluorine azide (FN3) is very explosive and thermally unstable. Dinitrogen difluoride (N2F2) exists as thermally interconvertible cis and trans isomers, and was first found as a product of the thermal decomposition of FN3.[57]
Nitrogen trichloride (NCl3) is a dense, volatile, and explosive liquid whose physical properties are similar to those of carbon tetrachloride, although one difference is that NCl3 is easily hydrolysed by water while CCl4 is not. It was first synthesised in 1811 by Pierre Louis Dulong, who lost three fingers and an eye to its explosive tendencies. As a dilute gas it is less dangerous and is thus used industrially to bleach and sterilise flour. Nitrogen tribromide (NBr3), first prepared in 1975, is a deep red, temperature-sensitive, volatile solid that is explosive even at −100 °C. Nitrogen triiodide (NI3) is still more unstable and was only prepared in 1990. Its adduct with ammonia, which was known earlier, is very shock-sensitive: it can be set off by the touch of a feather, shifting air currents, or even alpha particles.[57][58] For this reason, small amounts of nitrogen triiodide are sometimes synthesised as a demonstration to high school chemistry students or as an act of «chemical magic».[59] Chlorine azide (ClN3) and bromine azide (BrN3) are extremely sensitive and explosive.[60][61]
Two series of nitrogen oxohalides are known: the nitrosyl halides (XNO) and the nitryl halides (XNO2). The first is very reactive gases that can be made by directly halogenating nitrous oxide. Nitrosyl fluoride (NOF) is colourless and a vigorous fluorinating agent. Nitrosyl chloride (NOCl) behaves in much the same way and has often been used as an ionising solvent. Nitrosyl bromide (NOBr) is red. The reactions of the nitryl halides are mostly similar: nitryl fluoride (FNO2) and nitryl chloride (ClNO2) are likewise reactive gases and vigorous halogenating agents.[57]
Oxides
Nitrogen dioxide at −196 °C, 0 °C, 23 °C, 35 °C, and 50 °C. NO
2 converts to colourless dinitrogen tetroxide (N
2O
4) at low temperatures, and reverts to NO
2 at higher temperatures.
Nitrogen forms nine molecular oxides, some of which were the first gases to be identified: N2O (nitrous oxide), NO (nitric oxide), N2O3 (dinitrogen trioxide), NO2 (nitrogen dioxide), N2O4 (dinitrogen tetroxide), N2O5 (dinitrogen pentoxide), N4O (nitrosylazide),[62] and N(NO2)3 (trinitramide).[63] All are thermally unstable towards decomposition to their elements. One other possible oxide that has not yet been synthesised is oxatetrazole (N4O), an aromatic ring.[62]
Nitrous oxide (N2O), better known as laughing gas, is made by thermal decomposition of molten ammonium nitrate at 250 °C. This is a redox reaction and thus nitric oxide and nitrogen are also produced as byproducts. It is mostly used as a propellant and aerating agent for sprayed canned whipped cream, and was formerly commonly used as an anaesthetic. Despite appearances, it cannot be considered to be the anhydride of hyponitrous acid (H2N2O2) because that acid is not produced by the dissolution of nitrous oxide in water. It is rather unreactive (not reacting with the halogens, the alkali metals, or ozone at room temperature, although reactivity increases upon heating) and has the unsymmetrical structure N–N–O (N≡N+O−↔−N=N+=O): above 600 °C it dissociates by breaking the weaker N–O bond.[62]
Nitric oxide (NO) is the simplest stable molecule with an odd number of electrons. In mammals, including humans, it is an important cellular signaling molecule involved in many physiological and pathological processes.[64] It is formed by catalytic oxidation of ammonia. It is a colourless paramagnetic gas that, being thermodynamically unstable, decomposes to nitrogen and oxygen gas at 1100–1200 °C. Its bonding is similar to that in nitrogen, but one extra electron is added to a π* antibonding orbital and thus the bond order has been reduced to approximately 2.5; hence dimerisation to O=N–N=O is unfavourable except below the boiling point (where the cis isomer is more stable) because it does not actually increase the total bond order and because the unpaired electron is delocalised across the NO molecule, granting it stability. There is also evidence for the asymmetric red dimer O=N–O=N when nitric oxide is condensed with polar molecules. It reacts with oxygen to give brown nitrogen dioxide and with halogens to give nitrosyl halides. It also reacts with transition metal compounds to give nitrosyl complexes, most of which are deeply coloured.[62]
Blue dinitrogen trioxide (N2O3) is only available as a solid because it rapidly dissociates above its melting point to give nitric oxide, nitrogen dioxide (NO2), and dinitrogen tetroxide (N2O4). The latter two compounds are somewhat difficult to study individually because of the equilibrium between them, although sometimes dinitrogen tetroxide can react by heterolytic fission to nitrosonium and nitrate in a medium with high dielectric constant. Nitrogen dioxide is an acrid, corrosive brown gas. Both compounds may be easily prepared by decomposing a dry metal nitrate. Both react with water to form nitric acid. Dinitrogen tetroxide is very useful for the preparation of anhydrous metal nitrates and nitrato complexes, and it became the storable oxidiser of choice for many rockets in both the United States and USSR by the late 1950s. This is because it is a hypergolic propellant in combination with a hydrazine-based rocket fuel and can be easily stored since it is liquid at room temperature.[62]
The thermally unstable and very reactive dinitrogen pentoxide (N2O5) is the anhydride of nitric acid, and can be made from it by dehydration with phosphorus pentoxide. It is of interest for the preparation of explosives.[65] It is a deliquescent, colourless crystalline solid that is sensitive to light. In the solid state it is ionic with structure [NO2]+[NO3]−; as a gas and in solution it is molecular O2N–O–NO2. Hydration to nitric acid comes readily, as does analogous reaction with hydrogen peroxide giving peroxonitric acid (HOONO2). It is a violent oxidising agent. Gaseous dinitrogen pentoxide decomposes as follows:[62]
- N2O5 ⇌ NO2 + NO3 → NO2 + O2 + NO
- N2O5 + NO ⇌ 3 NO2
Oxoacids, oxoanions, and oxoacid salts
Many nitrogen oxoacids are known, though most of them are unstable as pure compounds and are known only as aqueous solutions or as salts. Hyponitrous acid (H2N2O2) is a weak diprotic acid with the structure HON=NOH (pKa1 6.9, pKa2 11.6). Acidic solutions are quite stable but above pH 4 base-catalysed decomposition occurs via [HONNO]− to nitrous oxide and the hydroxide anion. Hyponitrites (involving the N
2O2−
2 anion) are stable to reducing agents and more commonly act as reducing agents themselves. They are an intermediate step in the oxidation of ammonia to nitrite, which occurs in the nitrogen cycle. Hyponitrite can act as a bridging or chelating bidentate ligand.[66]
Nitrous acid (HNO2) is not known as a pure compound, but is a common component in gaseous equilibria and is an important aqueous reagent: its aqueous solutions may be made from acidifying cool aqueous nitrite (NO−
2, bent) solutions, although already at room temperature disproportionation to nitrate and nitric oxide is significant. It is a weak acid with pKa 3.35 at 18 °C. They may be titrimetrically analysed by their oxidation to nitrate by permanganate. They are readily reduced to nitrous oxide and nitric oxide by sulfur dioxide, to hyponitrous acid with tin(II), and to ammonia with hydrogen sulfide. Salts of hydrazinium N
2H+
5 react with nitrous acid to produce azides which further react to give nitrous oxide and nitrogen. Sodium nitrite is mildly toxic in concentrations above 100 mg/kg, but small amounts are often used to cure meat and as a preservative to avoid bacterial spoilage. It is also used to synthesise hydroxylamine and to diazotise primary aromatic amines as follows:[66]
- ArNH2 + HNO2 → [ArNN]Cl + 2 H2O
Nitrite is also a common ligand that can coordinate in five ways. The most common are nitro (bonded from the nitrogen) and nitrito (bonded from an oxygen). Nitro-nitrito isomerism is common, where the nitrito form is usually less stable.[66]
Fuming nitric acid contaminated with yellow nitrogen dioxide
Nitric acid (HNO3) is by far the most important and the most stable of the nitrogen oxoacids. It is one of the three most used acids (the other two being sulfuric acid and hydrochloric acid) and was first discovered by alchemists in the 13th century. It is made by the catalytic oxidation of ammonia to nitric oxide, which is oxidised to nitrogen dioxide, and then dissolved in water to give concentrated nitric acid. In the United States of America, over seven million tonnes of nitric acid are produced every year, most of which is used for nitrate production for fertilisers and explosives, among other uses. Anhydrous nitric acid may be made by distilling concentrated nitric acid with phosphorus pentoxide at low pressure in glass apparatus in the dark. It can only be made in the solid state, because upon melting it spontaneously decomposes to nitrogen dioxide, and liquid nitric acid undergoes self-ionisation to a larger extent than any other covalent liquid as follows:[66]
- 2 HNO3 ⇌ H
2NO+
3 + NO−
3 ⇌ H2O + [NO2]+ + [NO3]−
Two hydrates, HNO3·H2O and HNO3·3H2O, are known that can be crystallised. It is a strong acid and concentrated solutions are strong oxidising agents, though gold, platinum, rhodium, and iridium are immune to attack. A 3:1 mixture of concentrated hydrochloric acid and nitric acid, called aqua regia, is still stronger and successfully dissolves gold and platinum, because free chlorine and nitrosyl chloride are formed and chloride anions can form strong complexes. In concentrated sulfuric acid, nitric acid is protonated to form nitronium, which can act as an electrophile for aromatic nitration:[66]
- HNO3 + 2 H2SO4 ⇌ NO+
2 + H3O+ + 2 HSO−
4
The thermal stabilities of nitrates (involving the trigonal planar NO−
3 anion) depends on the basicity of the metal, and so do the products of decomposition (thermolysis), which can vary between the nitrite (for example, sodium), the oxide (potassium and lead), or even the metal itself (silver) depending on their relative stabilities. Nitrate is also a common ligand with many modes of coordination.[66]
Finally, although orthonitric acid (H3NO4), which would be analogous to orthophosphoric acid, does not exist, the tetrahedral orthonitrate anion NO3−
4 is known in its sodium and potassium salts:[66]
These white crystalline salts are very sensitive to water vapour and carbon dioxide in the air:[66]
- Na3NO4 + H2O + CO2 → NaNO3 + NaOH + NaHCO3
Despite its limited chemistry, the orthonitrate anion is interesting from a structural point of view due to its regular tetrahedral shape and the short N–O bond lengths, implying significant polar character to the bonding.[66]
Organic nitrogen compounds
Nitrogen is one of the most important elements in organic chemistry. Many organic functional groups involve a carbon–nitrogen bond, such as amides (RCONR2), amines (R3N), imines (RC(=NR)R), imides (RCO)2NR, azides (RN3), azo compounds (RN2R), cyanates and isocyanates (ROCN or RCNO), nitrates (RONO2), nitriles and isonitriles (RCN or RNC), nitrites (RONO), nitro compounds (RNO2), nitroso compounds (RNO), oximes (RCR=NOH), and pyridine derivatives. C–N bonds are strongly polarised towards nitrogen. In these compounds, nitrogen is usually trivalent (though it can be tetravalent in quaternary ammonium salts, R4N+), with a lone pair that can confer basicity on the compound by being coordinated to a proton. This may be offset by other factors: for example, amides are not basic because the lone pair is delocalised into a double bond (though they may act as acids at very low pH, being protonated at the oxygen), and pyrrole is not acidic because the lone pair is delocalised as part of an aromatic ring.[67] The amount of nitrogen in a chemical substance can be determined by the Kjeldahl method.[68] In particular, nitrogen is an essential component of nucleic acids, amino acids and thus proteins, and the energy-carrying molecule adenosine triphosphate and is thus vital to all life on Earth.[67]
Occurrence
Nitrogen is the most common pure element in the earth, making up 78.1% of the volume of the atmosphere[6] (75.5% by mass), around 3.89 million gigatonnes. Despite this, it is not very abundant in Earth’s crust, making up somewhere around 19 parts per million of this, on par with niobium, gallium, and lithium. (This represents 300,000 to a million gigatonnes of nitrogen, depending on the mass of the crust.[69]) The only important nitrogen minerals are nitre (potassium nitrate, saltpetre) and soda nitre (sodium nitrate, Chilean saltpetre). However, these have not been an important source of nitrates since the 1920s, when the industrial synthesis of ammonia and nitric acid became common.[70]
Nitrogen compounds constantly interchange between the atmosphere and living organisms. Nitrogen must first be processed, or «fixed», into a plant-usable form, usually ammonia. Some nitrogen fixation is done by lightning strikes producing the nitrogen oxides, but most is done by diazotrophic bacteria through enzymes known as nitrogenases (although today industrial nitrogen fixation to ammonia is also significant). When the ammonia is taken up by plants, it is used to synthesise proteins. These plants are then digested by animals who use the nitrogen compounds to synthesise their proteins and excrete nitrogen-bearing waste. Finally, these organisms die and decompose, undergoing bacterial and environmental oxidation and denitrification, returning free dinitrogen to the atmosphere. Industrial nitrogen fixation by the Haber process is mostly used as fertiliser, although excess nitrogen–bearing waste, when leached, leads to eutrophication of freshwater and the creation of marine dead zones, as nitrogen-driven bacterial growth depletes water oxygen to the point that all higher organisms die. Furthermore, nitrous oxide, which is produced during denitrification, attacks the atmospheric ozone layer.[70]
Many saltwater fish manufacture large amounts of trimethylamine oxide to protect them from the high osmotic effects of their environment; conversion of this compound to dimethylamine is responsible for the early odour in unfresh saltwater fish.[71] In animals, free radical nitric oxide (derived from an amino acid), serves as an important regulatory molecule for circulation.[72]
Nitric oxide’s rapid reaction with water in animals results in the production of its metabolite nitrite. Animal metabolism of nitrogen in proteins, in general, results in the excretion of urea, while animal metabolism of nucleic acids results in the excretion of urea and uric acid. The characteristic odour of animal flesh decay is caused by the creation of long-chain, nitrogen-containing amines, such as putrescine and cadaverine, which are breakdown products of the amino acids ornithine and lysine, respectively, in decaying proteins.[73]
Production
Nitrogen gas is an industrial gas produced by the fractional distillation of liquid air, or by mechanical means using gaseous air (pressurised reverse osmosis membrane or pressure swing adsorption). Nitrogen gas generators using membranes or pressure swing adsorption (PSA) are typically more cost and energy efficient than bulk-delivered nitrogen.[74] Commercial nitrogen is often a byproduct of air-processing for industrial concentration of oxygen for steelmaking and other purposes. When supplied compressed in cylinders it is often called OFN (oxygen-free nitrogen).[75] Commercial-grade nitrogen already contains at most 20 ppm oxygen, and specially purified grades containing at most 2 ppm oxygen and 10 ppm argon are also available.[76]
In a chemical laboratory, it is prepared by treating an aqueous solution of ammonium chloride with sodium nitrite.[77]
- NH4Cl + NaNO2 → N2 + NaCl + 2 H2O
Small amounts of the impurities NO and HNO3 are also formed in this reaction. The impurities can be removed by passing the gas through aqueous sulfuric acid containing potassium dichromate.[77] Very pure nitrogen can be prepared by the thermal decomposition of barium azide or sodium azide.[78]
- 2 NaN3 → 2 Na + 3 N2
Applications
Gas
The applications of nitrogen compounds are naturally extremely widely varied due to the huge size of this class: hence, only applications of pure nitrogen itself will be considered here. Two-thirds (2/3) of nitrogen produced by industry is sold as gas and the remaining one-third (1/3) as a liquid.
The gas is mostly used as a low reactivity safe atmosphere wherever the oxygen in the air would pose a fire, explosion, or oxidising hazard. Some examples include:[76]
- As a modified atmosphere, pure or mixed with carbon dioxide, to nitrogenate and preserve the freshness of packaged or bulk foods (by delaying rancidity and other forms of oxidative damage). Pure nitrogen as food additive is labeled in the European Union with the E number E941.[79]
- In incandescent light bulbs as an inexpensive alternative to argon.[80]
- In fire suppression systems for Information technology (IT) equipment.[76]
- In the manufacture of stainless steel.[81]
- In the case-hardening of steel by nitriding.[82]
- In some aircraft fuel systems to reduce fire hazard (see inerting system).
- To inflate race car and aircraft tires,[83] reducing the problems of inconsistent expansion and contraction caused by moisture and oxygen in natural air.[76]
Nitrogen is commonly used during sample preparation in chemical analysis. It is used to concentrate and reduce the volume of liquid samples. Directing a pressurised stream of nitrogen gas perpendicular to the surface of the liquid causes the solvent to evaporate while leaving the solute(s) and un-evaporated solvent behind.[84]
Nitrogen can be used as a replacement, or in combination with, carbon dioxide to pressurise kegs of some beers, particularly stouts and British ales, due to the smaller bubbles it produces, which makes the dispensed beer smoother and headier.[85] A pressure-sensitive nitrogen capsule known commonly as a «widget» allows nitrogen-charged beers to be packaged in cans and bottles.[86][87] Nitrogen tanks are also replacing carbon dioxide as the main power source for paintball guns. Nitrogen must be kept at a higher pressure than CO2, making N2 tanks heavier and more expensive.[88]
Equipment
Some construction equipment uses pressurized nitrogen gas to help hydraulic system to provide extra power to devices such as hydraulic hammer. Nitrogen gas, formed from the decomposition of sodium azide, is used for the inflation of airbags.[89]
Execution
As nitrogen is an asphyxiant gas, some jurisdictions have considered asphyxiation by inhalation of pure nitrogen as a means of capital punishment (as a substitute for lethal injection).[90][91][92]
However, as of 2020, no executions using nitrogen gas have yet been carried out by any jurisdiction, and at least one jurisdiction (Oklahoma) which had considered nitrogen asphyxiation as an execution protocol had abandoned the effort.[93]
Liquid
Air balloon submerged in liquid nitrogen
Liquid nitrogen is a cryogenic liquid which looks like water. When insulated in proper containers such as dewar flasks, it can be transported and stored with a low rate of evaporative loss.[94]
A container vehicle carrying liquid nitrogen.
Like dry ice, the main use of liquid nitrogen is for cooling to low temperatures. It is used in the cryopreservation of biological materials such as blood and reproductive cells (sperm and eggs). It is used in cryotherapy to remove cysts and warts on the skin by freezing them.[95] It is used in laboratory cold traps, and in cryopumps to obtain lower pressures in vacuum pumped systems. It is used to cool heat-sensitive electronics such as infrared detectors and X-ray detectors. Other uses include freeze-grinding and machining materials that are soft or rubbery at room temperature, shrink-fitting and assembling engineering components, and more generally to attain very low temperatures where necessary. Because of its low cost, liquid nitrogen is often used for cooling even when such low temperatures are not strictly necessary, such as refrigeration of food, freeze-branding livestock, freezing pipes to halt flow when valves are not present, and consolidating unstable soil by freezing whenever excavation is going on underneath.[76]
Safety
Gas
Although nitrogen is non-toxic, when released into an enclosed space it can displace oxygen, and therefore presents an asphyxiation hazard. This may happen with few warning symptoms, since the human carotid body is a relatively poor and slow low-oxygen (hypoxia) sensing system.[96] An example occurred shortly before the launch of the first Space Shuttle mission on March 19, 1981, when two technicians died from asphyxiation after they walked into a space located in the Space Shuttle’s mobile launcher platform that was pressurised with pure nitrogen as a precaution against fire.[97]
When inhaled at high partial pressures (more than about 4 bar, encountered at depths below about 30 m in scuba diving), nitrogen is an anesthetic agent, causing nitrogen narcosis, a temporary state of mental impairment similar to nitrous oxide intoxication.[98][99]
Nitrogen dissolves in the blood and body fats. Rapid decompression (as when divers ascend too quickly or astronauts decompress too quickly from cabin pressure to spacesuit pressure) can lead to a potentially fatal condition called decompression sickness (formerly known as caisson sickness or the bends), when nitrogen bubbles form in the bloodstream, nerves, joints, and other sensitive or vital areas.[100][101] Bubbles from other «inert» gases (gases other than carbon dioxide and oxygen) cause the same effects, so replacement of nitrogen in breathing gases may prevent nitrogen narcosis, but does not prevent decompression sickness.[102]
Liquid
As a cryogenic liquid, liquid nitrogen can be dangerous by causing cold burns on contact, although the Leidenfrost effect provides protection for very short exposure (about one second).[103] Ingestion of liquid nitrogen can cause severe internal damage. For example, in 2012, a young woman in England had to have her stomach removed after ingesting a cocktail made with liquid nitrogen.[104]
Because the liquid-to-gas expansion ratio of nitrogen is 1:694 at 20 °C, a tremendous amount of force can be generated if liquid nitrogen is rapidly vaporised in an enclosed space. In an incident on January 12, 2006, at Texas A&M University, the pressure-relief devices of a tank of liquid nitrogen were malfunctioning and later sealed. As a result of the subsequent pressure buildup, the tank failed catastrophically. The force of the explosion was sufficient to propel the tank through the ceiling immediately above it, shatter a reinforced concrete beam immediately below it, and blow the walls of the laboratory 0.1–0.2 m off their foundations.[105]
Liquid nitrogen readily evaporates to form gaseous nitrogen, and hence the precautions associated with gaseous nitrogen also apply to liquid nitrogen.[106][107][108] For example, oxygen sensors are sometimes used as a safety precaution when working with liquid nitrogen to alert workers of gas spills into a confined space.[109]
Vessels containing liquid nitrogen can condense oxygen from air. The liquid in such a vessel becomes increasingly enriched in oxygen (boiling point −183 °C, higher than that of nitrogen) as the nitrogen evaporates, and can cause violent oxidation of organic material.[110]
Oxygen deficiency monitors
Oxygen deficiency monitors are used to measure levels of oxygen in confined spaces and any place where nitrogen gas or liquid are stored or used. In the event of a nitrogen leak, and a decrease in oxygen to a pre-set alarm level, an oxygen deficiency monitor can be programmed to set off audible and visual alarms, thereby providing notification of the possible impending danger. Most commonly the oxygen range to alert personnel is when oxygen levels get below 19.5%. OSHA specifies that a hazardous atmosphere may include one where the oxygen concentration is below 19.5% or above 23.5%.[111]
Oxygen deficiency monitors can either be fixed, mounted to the wall and hard-wired into the building’s power supply or simply plugged into a power outlet, or a portable hand-held or wearable monitor.
See also
- Reactive nitrogen species
- Soil gas
References
- ^ «Standard Atomic Weights: Nitrogen». CIAAW. 2009.
- ^ a b c Lide, David R. (1990–1991). CRC Handbook of Physics and Chemistry (71st ed.). Boca Raton, Ann Arbor, Boston: CRC Press, inc. pp. 4-22 (one page).
- ^ «Gases — Density». The Engineering Toolbox. Retrieved 27 January 2019.
- ^ Tetrazoles contain a pair of double-bonded nitrogen atoms with oxidation state 0 in the ring. A Synthesis of the parent 1H-tetrazole, CH2N4 (two atoms N(0)) is given in Ronald A. Henry and William G. Finnegan, «An Improved Procedure for the Deamination of 5-Aminotetrazole», _J. Am. Chem. Soc._ (1954), 76, 1, 290–291,
https://doi.org/10.1021/ja01630a086. - ^ Common Bond Energies (D) and Bond Lengths (r) Archived 2010-05-15 at the Wayback Machine. wiredchemist.com
- ^ a b c Greenwood and Earnshaw, pp. 406–07
- ^ Rutherford, Daniel (1772) «Dissertatio Inauguralis de aere fixo, aut mephitico Archived 2020-08-06 at the Wayback Machine» (Inaugural dissertation on the air [called] fixed or mephitic), M.D. dissertation, University of Edinburgh, Scotland. English translation: Dobbin, Leonard (1935). «Daniel Rutherford’s inaugural dissertation». Journal of Chemical Education. 12 (8): 370–75. Bibcode:1935JChEd..12..370D. doi:10.1021/ed012p370.
- ^ Weeks, Mary Elvira (1932). «The discovery of the elements. IV. Three important gases». Journal of Chemical Education. 9 (2): 215. Bibcode:1932JChEd…9..215W. doi:10.1021/ed009p215.
- ^ Aaron J. Ihde, The Development of Modern Chemistry, New York 1964.
- ^ Carl Wilhelm Scheele, Chemische Abhandlung von der Luft und dem Feuer [Chemical treatise on air and fire] (Upsala, Sweden: Magnus Swederus, 1777; and Leipzig, (Germany): Siegfried Lebrecht Crusius, 1777). In the section titled «Die Luft muß aus elastischen Flüßigkeiten von zweyerley Art, zusammengesetzet seyn.» (The air must be composed of elastic fluids of two sorts), pp. 6–14, Scheele presents the results of eight experiments in which air was reacted with various substances. He concluded (p. 13): «So viel sehe ich aus angeführten Versuchen, daß die Luft aus 2 von einander unterschiedenen Flußigkeiten bestehe, von welchen die eine die Eigenschaft das Phlogiston anzuziehen gar nicht äussere, die andere aber zur solchen Attraction eigentlich aufgeleget ist und welche zwischen dem 3:ten und 4:ten Theil von der ganzen Luftmasse aus machet.» (So I see [this] much from the experiments [that were] conducted: that the air consists of two fluids [that] differ from one another, of which the one doesn’t express at all the property of attracting phlogiston; the other, however, is capable of such attraction and which makes up between 1/3 and 1/4 part of the entire mass of the air.)
- ^ Priestley, Joseph (1772). «Observations on different kinds of air». Philosophical Transactions of the Royal Society of London. 62: 147–256. doi:10.1098/rstl.1772.0021. S2CID 186210131. ; see p. 225. Archived 2016-09-03 at the Wayback Machine
- ^ Priestley, Joseph (1772). «Observations on different kinds of air». Philosophical Transactions of the Royal Society of London. 62: 147–256. doi:10.1098/rstl.1772.0021. S2CID 186210131. ; see: «VII. Of air infected with the fumes of burning charcoal.» pp. 225–28. Archived 2016-09-03 at the Wayback Machine
- ^ Lavoisier, Antoine with Robert Kerr, trans., Elements of Chemistry, 4th ed. (Edinburgh, Scotland: William Creech, 1799), pp. 85–86. [p. 85]: Archived 2020-08-06 at the Wayback Machine «In reflecting upon the circumstances of this experiment, we readily perceive; that the mercury, during its calcination [i.e., roasting in air], absorbs the salubrious and respirable part of the air, or, to speak more strictly, the base of this respirable part; that the remaining air is a species of mephitis [i.e., a poisonous gas emitted from the earth], incapable of supporting combustion or respiration; … » [p. 86]: Archived 2020-08-06 at the Wayback Machine «I shall afterwards shew, that at least in our climate, the atmospheric air is composed of respirable and mephitic airs, in the proportion of 27 and 73; … «
- ^ Lavoisier, Antoine with Robert Kerr, trans., Elements of Chemistry, 4th ed. (Edinburgh, Scotland: William Creech, 1799), p. 101: «The chemical properties of the noxious portion of the atmospheric air being hitherto but little known, we have been satisfied to derive the name of its base from its known quality of killing such animals as are forced to breathe it, giving it the name of azot, from the Greek privitive particle α and ξωη, vita; hence the name of the noxious part of atmospheric air is azotic gas.»
- ^ Chaptal, J. A. and Nicholson, William trans. (1800) Elements of Chemistry, 3rd ed. London, England: C.C. and J. Robinson, vol. 1. pp. xxxv–xxxvi: «In order to correct the Nomenclature on this head [i.e., in this regard], nothing more is necessary than to substitute to [i.e., for] this word a denomination which is derived from the general system made use of; and I have presumed to propose that of Nitrogene Gas. In the first place, it is deduced from the characteristic and exclusive property of this gas, which forms the radical of the nitric acid. By this means we shall preserve to the combinations [i.e., compounds] of this substance the received [i.e., prevailing] denominations, such as those of the Nitric Acid, Nitrates, Nitrites, &c.»
- ^ nitrogen Archived 2017-07-02 at the Wayback Machine. Etymonline.com. Retrieved 2011-10-26.
- ^ Strutt, R. J. (1911) «Bakerian Lecture. A chemically active modification of nitrogen, produced by the electric discharge,» Archived 2016-12-20 at the Wayback Machine Proceedings of the Royal Society A, 85 (577): 219–29.
- ^ Lord Rayleigh’s Active Nitrogen Archived 2012-11-01 at the Wayback Machine. Lateralscience.co.uk. Retrieved 2011-10-26.
- ^ Erisman, Jan Willem; Sutton, Mark A.; Galloway, James; Klimont, Zbigniew; Winiwarter, Wilfried (2008). «How a century of ammonia synthesis changed the world». Nature Geoscience. 1 (10): 636. Bibcode:2008NatGe…1..636E. doi:10.1038/ngeo325. S2CID 94880859.
- ^ GB 190200698, Ostwald, Wilhelm, «Improvements in the Manufacture of Nitric Acid and Nitrogen Oxides», published 1902-03-20
- ^ GB 190208300, Ostwald, Wilhelm, «Improvements in and relating to the Manufacture of Nitric Acid and Oxides of Nitrogen», published 1903-02-26
- ^ a b c d e Greenwood and Earnshaw, pp. 411–12
- ^ Greenwood and Earnshaw, p. 550
- ^ Kaupp, Martin (1 December 2006). «The role of radial nodes of atomic orbitals for chemical bonding and the periodic table». Journal of Computational Chemistry. 28 (1): 320–25. doi:10.1002/jcc.20522. PMID 17143872. S2CID 12677737.
- ^ a b c d e f g h i j Greenwood and Earnshaw, pp. 412–16
- ^ Miller, T. S.; Belen, A.; Suter, T. M.; Sella, A.; Corà, A.; McMillan, P. F. (2017). «Carbon nitrides: synthesis and characterization of a new class of functional materials». Physical Chemistry Chemical Physics. 19 (24): 15613–15638. Bibcode:2017PCCP…1915613M. doi:10.1039/C7CP02711G. PMID 28594419.
- ^ House, J. E.; House, K. A. (2016). Descriptive Inorganic Chemistry. Amsterdam: Elsevier. p. 198. ISBN 978-0-12-804697-5.
- ^ Roy, A. K.; Burns, G. T.; Grigora, S.; Lie, G. C. (1994). «Poly(alkyl/aryloxothiazenes), [N=S(O)R]n : New direction in inorganic polymers». In Wisian-Neilson, P.; Alcock, H. R.; Wynne, K. J. (eds.). Inorganic and organometallic polymers II: advanced materials and intermediates. American Chemical Society. pp. 344–357. doi:10.1021/bk-1994-0572.ch026.
- ^ Holman, Jack P. (2002). Heat transfer (9th ed.). New York, NY: McGraw-Hill Companies, Inc. pp. 600–606. ISBN 9780072406559. OCLC 46959719.
- ^ Incropera 1 Dewitt 2 Bergman 3 Lavigne 4, Frank P. 1 David P. 2 Theodore L. 3 Adrienne S. 4 (2007). Fundamentals of heat and mass transfer (6th ed.). Hoboken, NJ: John Wiley and Sons, Inc. pp. 941–950. ISBN 9780471457282. OCLC 62532755.
- ^ Bethe, H. A. (1939). «Energy Production in Stars». Physical Review. 55 (5): 434–56. Bibcode:1939PhRv…55..434B. doi:10.1103/PhysRev.55.434. PMID 17835673.
- ^ CIAAW (2003). «Atomic Weight of Nitrogen». ciaaw.org. CIAAW. Archived from the original on 14 October 2016. Retrieved 13 October 2016.
- ^ Flanagan, Lawrence B.; Ehleringer, James R.; Pataki, Diane E. (15 December 2004). Stable Isotopes and Biosphere – Atmosphere Interactions: Processes and Biological Controls. pp. 74–75. ISBN 978-0-08-052528-0. Archived from the original on 5 February 2016. Retrieved 20 December 2015.
- ^ Greenwood and Earnshaw, p. 408
- ^ «Evaluated Nuclear Data File (ENDF) Retrieval & Plotting». National Nuclear Data Center. Archived from the original on 2020-08-09. Retrieved 2016-11-23.
- ^ Arthur G Palmer (2007). Protein NMR Spectroscopy. Elsevier Academic Press. ISBN 978-0-12-164491-8.
- ^ Katzenberg, M. A. (2008). «Chapter 13: Stable Isotope Analysis: A Tool for Studying Past Diet, Demography, and Life History». Biological Anthropology of the Human Skeleton (2nd ed.). ISBN 978-0-471-79372-4.
- ^ a b c Audi, Georges; Bersillon, Olivier; Blachot, Jean; Wapstra, Aaldert Hendrik (2003), «The NUBASE evaluation of nuclear and decay properties», Nuclear Physics A, 729: 3–128, Bibcode:2003NuPhA.729….3A, doi:10.1016/j.nuclphysa.2003.11.001
- ^ Carlson, Neil (January 22, 2012). Physiology of Behavior. Methods and Strategies of Research. Vol. 11th edition. Pearson. p. 151. ISBN 978-0-205-23939-9.
- ^ a b c Neeb, Karl Heinz (1997). The Radiochemistry of Nuclear Power Plants with Light Water Reactors. Berlin-New York: Walter de Gruyter. p. 227. ISBN 978-3-11-013242-7. Archived from the original on 2016-02-05. Retrieved 2015-12-20.
- ^ «Universal Industrial Gases, Inc…Nitrogen N2 Properties, Uses, Applications — Gas and Liquid».
- ^ Lewars, Errol G. (2008). Modeling Marvels: Computational Anticipation of Novel molecules. Springer Science+Business Media. pp. 141–63. doi:10.1007/978-1-4020-6973-4. ISBN 978-1-4020-6972-7.
- ^ «Polymeric nitrogen synthesized». physorg.com. 5 August 2004. Archived from the original on 2012-01-24. Retrieved 2009-06-22.
- ^ Gray, Theodore (2009). The Elements: A Visual Exploration of Every Known Atom in the Universe. New York: Black Dog & Leventhal Publishers. ISBN 978-1-57912-814-2.
- ^ Schuch, A. F.; Mills, R. L. (1970). «Crystal Structures of the Three Modifications of Nitrogen 14 and Nitrogen 15 at High Pressure». The Journal of Chemical Physics. 52 (12): 6000–08. Bibcode:1970JChPh..52.6000S. doi:10.1063/1.1672899.
- ^ Iancu, C. V.; Wright, E. R.; Heymann, J. B.; Jensen, G. J. (2006). «A comparison of liquid nitrogen and liquid helium as cryogens for electron cryotomography». Journal of Structural Biology. 153 (3): 231–40. doi:10.1016/j.jsb.2005.12.004. PMID 16427786.
- ^ «Flowing nitrogen ice glaciers seen on surface of Pluto after New Horizons flyby». ABC News. 25 July 2015. Archived from the original on 29 September 2015. Retrieved 6 October 2015.
- ^ McKinnon, William B.; Kirk, Randolph L. (2014). «Triton». In Spohn, Tilman; Breuer, Doris; Johnson, Torrence (eds.). Encyclopedia of the Solar System (3rd ed.). Amsterdam; Boston: Elsevier. pp. 861–82. ISBN 978-0-12-416034-7. Archived from the original on 2016-09-03. Retrieved 2016-04-30.
- ^ «Neptune: Moons: Triton». NASA. Archived from the original on October 15, 2011. Retrieved September 21, 2007.
- ^ Fryzuk, M. D. & Johnson, S. A. (2000). «The continuing story of dinitrogen activation». Coordination Chemistry Reviews. 200–202: 379. doi:10.1016/S0010-8545(00)00264-2.
- ^ Schrock, R. R. (2005). «Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center». Acc. Chem. Res. 38 (12): 955–62. doi:10.1021/ar0501121. PMC 2551323. PMID 16359167.
- ^ a b c d e Greenwood and Earnshaw, pp. 417–20
- ^ Greenwood and Earnshaw, pp. 434–38
- ^ Greenwood and Earnshaw, pp. 420–26
- ^ a b c d Greenwood and Earnshaw, pp. 426–33
- ^ Vieira, R.; C. Pham-Huu; N. Keller; M. J. Ledoux (2002). «New carbon nanofiber/graphite felt composite for use as a catalyst for hydrazine catalytic decomposition». Chemical Communications (9): 954–55. doi:10.1039/b202032g. PMID 12123065.
- ^ a b c d Greenwood and Earnshaw, pp. 438–42
- ^ Bowden, F. P. (1958). «Initiation of Explosion by Neutrons, α-Particles, and Fission Products». Proceedings of the Royal Society of London A. 246 (1245): 216–19. Bibcode:1958RSPSA.246..216B. doi:10.1098/rspa.1958.0123. S2CID 137728239.
- ^ Ford, L. A.; Grundmeier, E. W. (1993). Chemical Magic. Dover. p. 76. ISBN 978-0-486-67628-9.
- ^ Frierson, W. J.; Kronrad, J.; Browne, A. W. (1943). «Chlorine Azide, ClN3. I». Journal of the American Chemical Society. 65 (9): 1696–1698. doi:10.1021/ja01249a012.
- ^ Lyhs, Benjamin; Bläser, Dieter; Wölper, Christoph; Schulz, Stephan; Jansen, Georg (20 February 2012). «Solid-State Structure of Bromine Azide» (PDF). Angewandte Chemie International Edition. 51 (8): 1970–1974. doi:10.1002/anie.201108092. PMID 22250068. Archived (PDF) from the original on 25 August 2021. Retrieved 25 August 2021.
- ^ a b c d e f Greenwood and Earnshaw, pp. 443–58
- ^ Rahm, Martin; Dvinskikh, Sergey V.; Furó, István; Brinck, Tore (23 December 2010). «Experimental Detection of Trinitramide, N(NO2)3«. Angewandte Chemie International Edition. 50 (5): 1145–48. doi:10.1002/anie.201007047. PMID 21268214. S2CID 32952729.
- ^ Hou, Y. C.; Janczuk, A.; Wang, P. G. (1999). «Current trends in the development of nitric oxide donors». Current Pharmaceutical Design. 5 (6): 417–41. PMID 10390607.
- ^ Talawar, M. B.; et al. (2005). «Establishment of Process Technology for the Manufacture of Dinitrogen Pentoxide and its Utility for the Synthesis of Most Powerful Explosive of Today – CL-20». Journal of Hazardous Materials. 124 (1–3): 153–64. doi:10.1016/j.jhazmat.2005.04.021. PMID 15979786.
- ^ a b c d e f g h i Greenwood and Earnshaw, pp. 459–72
- ^ a b March, Jerry (1985), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (3rd ed.), New York: Wiley, ISBN 0-471-85472-7
- ^ Rédei, George P (2008). «Kjeldahl Method». Encyclopedia of Genetics, Genomics, Proteomics and Informatics. p. 1063. doi:10.1007/978-1-4020-6754-9_9066. ISBN 978-1-4020-6753-2.
- ^ Depending on the average thickness which is somewhere between 10 and 30 km, the mass of the earth’s crust is between 15×1018 and 45×1018 tonnes.
- ^ a b Greenwood and Earnshaw, pp. 407–09
- ^ Nielsen, M. K.; Jørgensen, B. M. (Jun 2004). «Quantitative relationship between trimethylamine oxide aldolase activity and formaldehyde accumulation in white muscle from gadiform fish during frozen storage». Journal of Agricultural and Food Chemistry. 52 (12): 3814–22. doi:10.1021/jf035169l. PMID 15186102.
- ^ Knox, G. A. (2007). Biology of the Southern Ocean. CRC Press. p. 392. ISBN 978-0-8493-3394-1. Archived from the original on 2021-10-01. Retrieved 2020-08-24.
- ^ Vickerstaff Joneja; Janice M. (2004). Digestion, diet, and disease: irritable bowel syndrome and gastrointestinal function. Rutgers University Press. p. 121. ISBN 978-0-8135-3387-2. Archived from the original on 2021-10-01. Retrieved 2020-08-24.
- ^ Froehlich, Peter (May 2013). «A Sustainable Approach to the Supply of Nitrogen». www.parker.com. Parker Hannifin Corporation. Archived from the original on 16 March 2016. Retrieved 24 November 2016.
- ^ Reich, Murray; Kapenekas, Harry (1957). «Nitrogen Purfication. Pilot Plant Removal of Oxygen». Industrial & Engineering Chemistry. 49 (5): 869–73. doi:10.1021/ie50569a032.
- ^ a b c d e Greenwood and Earnshaw, pp. 409–11
- ^ a b Bartlett, J. K. (1967). «Analysis for nitrite by evolution of nitrogen: A general chemistry laboratory experiment». Journal of Chemical Education. 44 (8): 475. Bibcode:1967JChEd..44..475B. doi:10.1021/ed044p475.
- ^ Eremets, M. I.; Popov, M. Y.; Trojan, I. A.; Denisov, V. N.; Boehler, R.; Hemley, R. J. (2004). «Polymerization of nitrogen in sodium azide». The Journal of Chemical Physics. 120 (22): 10618–23. Bibcode:2004JChPh.12010618E. doi:10.1063/1.1718250. PMID 15268087.
- ^ Ministers, Nordic Council of (2002). Food Additives in Europe 2000. p. 591. ISBN 978-92-893-0829-8. Archived from the original on 2016-02-05. Retrieved 2015-12-20.
- ^ Harding, Charlie, ed. (2002). Elements of the p Block. Cambridge: Royal Society of Chemistry. ISBN 978-0-85404-690-4. Archived from the original on 2021-10-01. Retrieved 2020-08-24.
- ^ Gavriliuk, V. G.; Berns, Hans (1999). High nitrogen steels: structure, properties, manufacture, applications. Springer. ISBN 978-3-540-66411-6. Archived from the original on 2021-10-01. Retrieved 2020-08-24.
- ^ Meka, S. R.; Chauhan, A.; Steiner, T.; Bischoff, E.; Ghosh, P. K.; Mittemeijer, E. J. (2015). «Generating duplex microstructures by nitriding; nitriding of iron based Fe–Mn alloy». Materials Science and Technology. 32 (9): 1743284715Y.000. doi:10.1179/1743284715Y.0000000098.
- ^ «Why don’t they use normal air in race car tires?». Howstuffworks. 2001-03-16. Archived from the original on 2011-07-12. Retrieved 2006-07-22.
- ^ Kemmochi, Y; Tsutsumi, K.; Arikawa, A.; Nakazawa, H. (2002). «Centrifugal concentrator for the substitution of nitrogen blow-down micro-concentration in dioxin/polychlorinated biphenyl sample preparation». Journal of Chromatography A. 943 (2): 295–97. doi:10.1016/S0021-9673(01)01466-2. PMID 11833649.
- ^ Baxter, E. Denise; Hughes, Paul S. (2001). Beer: Quality, Safety and Nutritional Aspects. Royal Society of Chemistry. p. 22. ISBN 978-0-85404-588-4. Archived from the original on 2020-03-21. Retrieved 2015-06-20.
- ^ «How does the widget in a beer can work?». Howstuffworks. 2000-08-16. Archived from the original on 2007-11-02. Retrieved 2008-07-30.
- ^ Denny, Mark (1 November 2009). Froth!: The Science of Beer. p. 131. ISBN 978-0-8018-9569-2. Archived from the original on 5 February 2016. Retrieved 20 December 2015.
- ^ Kennett, Andrew J. (2008). Design of a pneumatically assisted shifting system for Formula SAE® racing applications (Thesis). Dept. of Mechanical Engineering, Massachusetts Institute of Technology. hdl:1721.1/45820.
- ^ Betterton, E. A. (2003). «Environmental Fate of Sodium Azide Derived from Automobile Airbags». Critical Reviews in Environmental Science and Technology. 33 (4): 423–58. doi:10.1080/10643380390245002. S2CID 96404307.
- ^ Sanburn, Josh (2015-04-10). «The Dawn of a New Form of Capital Punishment». Time. Archived from the original on 2015-04-11. Retrieved 2015-04-11.
- ^ Sexton, Mike (18 December 2012). «Euthanasia campaigner under scrutiny». ABC. Archived from the original on 7 July 2013. Retrieved 6 May 2013.
- ^ Berman, Mark (April 17, 2015). «Oklahoma says it will now use nitrogen gas as its backup method of execution». The Washington Post. Archived from the original on June 23, 2019. Retrieved June 22, 2019.
- ^ «Oklahoma Attorney general says state will resume executions». New York Post. Archived from the original on March 9, 2021. Retrieved March 22, 2020.
- ^ Kaganer, M. G.; Kozheurov, V. & Levina, Zh. L. (1967). «Vessels for the storage and transport of liquid oxygen and nitrogen». Chemical and Petroleum Engineering. 3 (12): 918–22. doi:10.1007/BF01136404. S2CID 96762552.
- ^ Ahmed I; Agarwal S; Ilchyshyn A; Charles-Holmes S; Berth-Jones J (May 2001). «Liquid nitrogen cryotherapy of common warts: cryo-spray vs. cotton wool bud». Br. J. Dermatol. 144 (5): 1006–09. doi:10.1046/j.1365-2133.2001.04190.x. PMID 11359389. S2CID 221325640.
- ^ «Biology Safety – Cryogenic materials. The risks posed by them». University of Bath. Archived from the original on February 6, 2007. Retrieved 2007-01-03.
- ^ «Space Shuttle Columbia Fast Facts». CNN. September 30, 2013. Archived from the original on February 2, 2016. Retrieved January 20, 2016.
- ^ Fowler, B.; Ackles, K. N.; Porlier, G. (1985). «Effects of inert gas narcosis on behavior – a critical review». Undersea Biomed. Res. 12 (4): 369–402. PMID 4082343. Archived from the original on 2010-12-25. Retrieved 2008-09-21.
{{cite journal}}: CS1 maint: unfit URL (link) - ^ Rogers, W. H.; Moeller, G. (1989). «Effect of brief, repeated hyperbaric exposures on susceptibility to nitrogen narcosis». Undersea Biomed. Res. 16 (3): 227–32. OCLC 2068005. PMID 2741255. Archived from the original on 2009-09-01. Retrieved 2008-09-21.
{{cite journal}}: CS1 maint: unfit URL (link) - ^ Acott, C. (1999). «A brief history of diving and decompression illness». South Pacific Underwater Medicine Society Journal. 29 (2). OCLC 16986801. Archived from the original on 2011-09-05. Retrieved 2008-09-21.
{{cite journal}}: CS1 maint: unfit URL (link) - ^ Kindwall, E. P.; Baz, A.; Lightfoot, E. N.; Lanphier, E. H.; Seireg, A. (1975). «Nitrogen elimination in man during decompression». Undersea Biomed. Res. 2 (4): 285–97. OCLC 2068005. PMID 1226586. Archived from the original on 2011-07-27. Retrieved 2008-09-21.
{{cite journal}}: CS1 maint: unfit URL (link) - ^ US Navy Diving Manual, 6th revision. United States: US Naval Sea Systems Command. 2006. Archived from the original on 2008-05-02. Retrieved 2008-04-24.
- ^ Walker, Jearl. «Boiling and the Leidenfrost Effect» (PDF). Fundamentals of Physics: 1–4. Archived (PDF) from the original on 13 December 2019. Retrieved 11 October 2014.
- ^ Liquid nitrogen cocktail leaves teen in hospital Archived 2017-04-12 at the Wayback Machine, BBC News, October 8, 2012.
- ^ Mattox, Brent S. «Investigative Report on Chemistry 301A Cylinder Explosion» (PDF). Texas A&M University. Archived from the original (reprint) on 2014-04-30.
- ^ British Compressed Gases Association (2000) BCGA Code of Practice CP30. The Safe Use of Liquid nitrogen Dewars up to 50 litres. Archived 2007-07-18 at the Wayback Machine ISSN 0260-4809.
- ^ Confined Space Entry – Worker and Would-be Rescuer Asphyxiated Archived 2015-09-22 at the Wayback Machine, Valero Refinery Asphyxiation Incident Case Study.
- ^ Inquiry after man dies in chemical leak Archived 2017-01-07 at the Wayback Machine, BBC News, October 25, 1999.
- ^ Liquid Nitrogen – Code of practice for handling. United Kingdom: Birkbeck, University of London. 2007. Archived from the original on 2018-06-12. Retrieved 2012-02-08.
- ^ Levey, Christopher G. «Liquid Nitrogen Safety». Thayer School of Engineering at Dartmouth. Archived from the original on 2016-03-05. Retrieved 2016-11-23.
- ^ National Institutes of Health. Protocol for Use and Maintenance of Oxygen Monitoring Devices. February 2014, at 1:35 UTC. Available at: https://www.ors.od.nih.gov/sr/dohs/documents/protocoloxygenmonitoring.pdf Archived 2020-12-05 at the Wayback Machine. Accessed June 23, 2020
Bibliography
- Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 978-0-08-037941-8.
External links
- Etymology of Nitrogen
- Nitrogen at The Periodic Table of Videos (University of Nottingham)
- Nitrogen podcast from the Royal Society of Chemistry’s Chemistry World
У этого термина существуют и другие значения, см. Азот (значения).
| |||
| Внешний вид простого вещества | |||
|---|---|---|---|
|
| |||
| Свойства атома | |||
| Имя, символ, номер | Азот / Nitrogenium (N), 7 | ||
| Атомная масса (молярная масса) | 14,00674 а. е. м. (г/моль) | ||
| Электронная конфигурация | [He] 2s2 2p3 | ||
| Радиус атома | 92 пм | ||
| Химические свойства | |||
| Ковалентный радиус | 75 пм | ||
| Радиус иона | 13 (+5e) 171 (-3e) пм | ||
| Электроотрицательность | 3,04[1] (шкала Полинга) | ||
| Степени окисления | 5, 4, 3, 2, 1, 0, −1, −3 | ||
| Энергия ионизации (первый электрон) | 1401,5 (14,53) кДж/моль (эВ) | ||
| Термодинамические свойства простого вещества | |||
| Плотность (при н. у.) | 0,808 г/см³ (−195,8 °C); при н.у. 0,001251 г/см³ | ||
| Температура плавления | 63,29 К (-209,86 °C) | ||
| Температура кипения | 77,4 К (-195,75 °C) | ||
| Теплота плавления | (N2) 0,720 кДж/моль | ||
| Теплота испарения | (N2) 5,57 кДж/моль | ||
| Молярная теплоёмкость | 29,125[2] (газ N2) Дж/(K·моль) | ||
| Молярный объём | 17,3 см³/моль | ||
| Кристаллическая решётка простого вещества | |||
| Структура решётки | кубическая | ||
| Параметры решётки | 5,661 Å | ||
| Прочие характеристики | |||
| Теплопроводность | (300 K) 0,026 Вт/(м·К) |

Азо́т — элемент 15-й группы (по устаревшей классификации — главной подгруппы пятой группы) второго периода периодической системы химических элементов Д. И. Менделеева, с атомным номером 7. Обозначается символом N (лат. Nitrogenium). Простое вещество азот (CAS-номер: 7727-37-9) — достаточно инертный при нормальных условиях двухатомный газ без цвета, вкуса и запаха (формула N2), из которого на три четверти состоит земная атмосфера.
Содержание
- 1 История открытия
- 2 Происхождение названия
- 3 Азот в природе
- 3.1 Изотопы
- 3.2 Распространённость
- 3.3 Биологическая роль
- 3.4 Круговорот азота в природе
- 3.5 Токсикология азота и его соединений
- 4 Получение
- 5 Свойства
- 5.1 Физические свойства
- 5.2 Химические свойства, строение молекулы
- 5.2.1 Промышленное связывание атмосферного азота
- 6 Соединения азота
- 7 Применение
- 8 Маркировка баллонов
- 9 Интересные факты
- 10 См. также
- 11 Примечания
- 12 Литература
- 13 Ссылки
История открытия
В 1772 году Генри Кавендиш провёл следующий опыт: он многократно пропускал воздух над раскалённым углём, затем обрабатывал его щёлочью, в результате получался остаток, который Кавендиш назвал удушливым (или мефитическим) воздухом. С позиций современной химии ясно, что в реакции с раскалённым углём кислород воздуха связывался в углекислый газ, который затем поглощался щёлочью. При этом остаток газа представлял собой по большей части азот. Таким образом, Кавендиш выделил азот, но не сумел понять, что это новое простое вещество (химический элемент). В том же году Кавендиш сообщил об этом опыте Джозефу Пристли.
Пристли в это время проводил серию экспериментов, в которых также связывал кислород воздуха и удалял полученный углекислый газ, то есть также получал азот, однако, будучи сторонником господствующей в те времена теории флогистона, совершенно неверно истолковал полученные результаты (по его мнению, процесс был противоположным — не кислород удалялся из газовой смеси, а наоборот, в результате обжига воздух насыщался флогистоном; оставшийся воздух (азот) он и назвал насыщенным флогистоном, то есть флогистированным). Очевидно, что и Пристли, хотя и смог выделить азот, не сумел понять сути своего открытия, поэтому и не считается первооткрывателем азота.
Одновременно схожие эксперименты с тем же результатом проводил и Карл Шееле.
В 1772 году азот (под названием «испорченного воздуха») как простое вещество описал Даниэль Резерфорд, он опубликовал магистерскую диссертацию, где указал основные свойства азота (не реагирует со щелочами, не поддерживает горения, непригоден для дыхания). Именно Даниэль Резерфорд и считается первооткрывателем азота. Однако и Резерфорд был сторонником флогистонной теории, поэтому также не смог понять, что же он выделил. Таким образом, чётко определить первооткрывателя азота невозможно.
В дальнейшем азот был изучен Генри Кавендишем (интересен тот факт, что он сумел связать азот с кислородом при помощи разрядов электрического тока, а после поглощения оксидов азота в остатке получил небольшое количество газа, абсолютно инертного, хотя, как и в случае с азотом, не смог понять, что выделил новый химический элемент — инертный газ аргон).
Происхождение названия
Азо́т (от др.-греч. ἄζωτος — безжизненный, лат. nitrogenium), вместо предыдущих названий («флогистированный», «мефитический» и «испорченный» воздух) предложил в 1787 году Антуан Лавуазье, который в то время в составе группы других французских учёных разрабатывал принципы химической номенклатуры. Как показано выше, в то время уже было известно, что азот не поддерживает ни горения, ни дыхания. Это свойство и сочли наиболее важным. Хотя впоследствии выяснилось, что азот, наоборот, крайне необходим для всех живых существ, название сохранилось во французском и русском языках.
Существует и иная версия[3]. Слово «азот» придумано не Лавуазье и не его коллегами по номенклатурной комиссии; оно вошло в алхимическую литературу уже в раннем средневековье и употреблялось для обозначения «первичной материи металлов», которую считали «альфой и омегой» всего сущего. Это выражение заимствовано из Апокалипсиса: «Я есмь Альфа и Омега, начало и конец» (Откр.1:8-10). Слово составлено из начальных и конечных букв алфавитов трёх языков — латинского, греческого и древнееврейского, — считавшихся «священными», поскольку, согласно Евангелиям, надпись на кресте при распятии Христа была сделана на этих языках (а, альфа, алеф и зет, омега, тав — AAAZOTH). Составители новой химической номенклатуры хорошо знали о существовании этого слова; инициатор её создания Гитон де Морво отмечал в своей «Методической энциклопедии» (1786) алхимическое значение термина.
Возможно, слово «азот» произошло от одного из двух арабских слов — либо от слова «аз-зат» («сущность» или «внутреннюю реальность»), либо от слова «зибак» («ртуть»)..
На латыни азот называется «nitrogenium», то есть «рождающий селитру»; английское название производится от латинского. В немецком языке используется название Stickstoff, что означает «удушающее вещество».
Азот в природе
Изотопы
Природный азот состоит из двух стабильных изотопов 14N — 99,635 % и 15N — 0,365 %.
Искусственно получены четырнадцать радиоактивных изотопов азота с массовыми числами от 10 до 13 и от 16 до 25. Все они являются очень короткоживущими изотопами. Самый стабильный из них 13N имеет период полураспада 10 мин.
Спин ядер стабильных изотопов азота: 14N — 1; 15N — 1/2.
Распространённость
Вне пределов Земли азот обнаружен в газовых туманностях, солнечной атмосфере, на Уране, Нептуне, межзвёздном пространстве и др. Азот — четвёртый по распространённости элемент Солнечной системы (после водорода, гелия и кислорода).
Азот, в форме двухатомных молекул N2 составляет большую часть атмосферы, где его содержание составляет 75,6 % (по массе) или 78,084 % (по объёму), то есть около 3,87·1015 т.
Содержание азота в земной коре, по данным разных авторов, составляет (0,7—1,5)·1015 т (причём в гумусе — порядка 6·1010 т), а в мантии Земли — 1,3·1016 т. Такое соотношение масс заставляет предположить, что главным источником азота служит верхняя часть мантии, откуда он поступает в другие оболочки Земли с извержениями вулканов.
Масса растворённого в гидросфере азота, учитывая, что одновременно происходят процессы растворения азота атмосферы в воде и выделения его в атмосферу, составляет около 2·1013 т, кроме того примерно 7·1011 т азота содержатся в гидросфере в виде соединений.
Биологическая роль
Азот является элементом, необходимым для существования животных и растений, он входит в состав белков (16—18 % по массе), аминокислот, нуклеиновых кислот, нуклеопротеидов, хлорофилла, гемоглобина и др. В составе живых клеток по числу атомов азота около 2 %, по массовой доле — около 2,5 % (четвёртое место после водорода, углерода и кислорода). В связи с этим значительное количество связанного азота содержится в живых организмах, «мёртвой органике» и дисперсном веществе морей и океанов. Это количество оценивается примерно в 1,9·1011 т. В результате процессов гниения и разложения азотсодержащей органики, при условии благоприятных факторов окружающей среды, могут образоваться природные залежи полезных ископаемых, содержащие азот, например, «чилийская селитра» (нитрат натрия с примесями других соединений), норвежская, индийская селитры.
Круговорот азота в природе
Фиксация атмосферного азота в природе происходит по двум основным направлениям — абиогенному и биогенному. Первый путь включает главным образом реакции азота с кислородом. Так как азот химически весьма инертен, для окисления требуются большие количества энергии (высокие температуры). Эти условия достигаются при разрядах молний, когда температура достигает 25000 °C и более. При этом происходит образование различных оксидов азота. Существует также вероятность, что абиотическая фиксация происходит в результате фотокаталитических реакций на поверхности полупроводников или широкополосных диэлектриков (песок пустынь).
Однако основная часть молекулярного азота (около 1,4·108 т/год) фиксируется биотическим путём. Долгое время считалось, что связывать молекулярный азот могут только небольшое количество видов микроорганизмов (хотя и широко распространённых на поверхности Земли): бактерии Azotobacter и Clostridium, клубеньковые бактерии бобовых растений Rhizobium, цианобактерии Anabaena, Nostoc и др. Сейчас известно, что этой способностью обладают многие другие организмы в воде и почве, например, актиномицеты в клубнях ольхи и других деревьев (всего 160 видов). Все они превращают молекулярный азот в соединения аммония (NH4+). Этот процесс требует значительных затрат энергии (для фиксации 1 г атмосферного азота бактерии в клубеньках бобовых расходуют порядка 167,5 кДж, то есть окисляют примерно 10 г глюкозы). Таким образом, видна взаимная польза от симбиоза растений и азотфиксирующих бактерий — первые предоставляют вторым «место для проживания» и снабжают полученным в результате фотосинтеза «топливом» — глюкозой, вторые обеспечивают необходимый растениям азот в усваиваемой ими форме.
Азот в форме аммиака и соединений аммония, получающийся в процессах биогенной азотфиксации, быстро окисляется до нитратов и нитритов (этот процесс носит название нитрификации). Последние, не связанные тканями растений (и далее по пищевой цепи травоядными и хищниками), недолго остаются в почве. Большинство нитратов и нитритов хорошо растворимы, поэтому они смываются водой и в конце концов попадают в мировой океан (этот поток оценивается в 2,5—8·107 т/год).
Азот, включённый в ткани растений и животных, после их гибели подвергается аммонификации (разложению содержащих азот сложных соединений с выделением аммиака и ионов аммония) и денитрификации, то есть выделению атомарного азота, а также его оксидов. Эти процессы целиком происходят благодаря деятельности микроорганизмов в аэробных и анаэробных условиях.
В отсутствие деятельности человека процессы связывания азота и нитрификации практически полностью уравновешены противоположными реакциями денитрификации. Часть азота поступает в атмосферу из мантии с извержениями вулканов, часть прочно фиксируется в почвах и глинистых минералах, кроме того, постоянно идёт утечка азота из верхних слоёв атмосферы в межпланетное пространство.
Токсикология азота и его соединений
Сам по себе атмосферный азот достаточно инертен, чтобы оказывать непосредственное влияние на организм человека и млекопитающих. Тем не менее, при повышенном давлении он вызывает наркоз, опьянение или удушье (при недостатке кислорода); при быстром снижении давления азот вызывает кессонную болезнь.
Многие соединения азота очень активны и нередко токсичны.
Получение
В лабораториях его можно получать по реакции разложения нитрита аммония:

Реакция экзотермическая, идёт с выделением 80 ккал (335 кДж), поэтому требуется охлаждение сосуда при её протекании (хотя для начала реакции требуется нагревание нитрита аммония).
Практически эту реакцию выполняют, добавляя по каплям насыщенный раствор нитрита натрия в нагретый насыщенный раствор сульфата аммония, при этом образующийся в результате обменной реакции нитрит аммония мгновенно разлагается.
Выделяющийся при этом газ загрязнён аммиаком, оксидом азота (I) и кислородом, от которых его очищают, последовательно пропуская через растворы серной кислоты, сульфата железа (II) и над раскалённой медью. Затем азот осушают.
Ещё один лабораторный способ получения азота — нагревание смеси дихромата калия и сульфата аммония (в соотношении 2:1 по массе). Реакция идёт по уравнениям:


Наиболее чистый азот можно получить разложением азидов металлов:
![mathsf{2NaN_3 xrightarrow[]{^ot} 2Na + 3N_2 uparrow}](https://dic.academic.ru/dic.nsf/ruwiki/c76a2446f01b021b5f88a9ac8e523bb5.png)
Так называемый «воздушный», или «атмосферный» азот, то есть смесь азота с благородными газами, получают путём реакции воздуха с раскалённым коксом, при этом образуется так называемый «генераторный», или «воздушный», газ — сырьё для химических синтезов и топливо. При необходимости из него можно выделить азот, поглотив монооксид углерода.
Молекулярный азот в промышленности получают фракционной перегонкой жидкого воздуха. Этим методом можно получить и «атмосферный азот». Также широко применяются азотные установки и станции, в которых используется метод адсорбционного и мембранного газоразделения.
Один из лабораторных способов — пропускание аммиака над оксидом меди (II) при температуре ~700 °C:

Аммиак берут из его насыщенного раствора при нагревании. Количество CuO в 2 раза больше расчётного. Непосредственно перед применением азот очищают от примеси кислорода и аммиака пропусканием над медью и её оксидом (II) (тоже ~700 °C), затем сушат концентрированной серной кислотой и сухой щёлочью. Процесс происходит довольно медленно, но он того стоит: газ получается весьма чистый.
Свойства
Физические свойства
![]()
![]()
Оптический линейчатый эмиссионный спектр азота
При нормальных условиях азот это бесцветный газ, не имеет запаха, мало растворим в воде (2,3 мл/100г при 0 °C, 0,8 мл/100 г при 80 °C), плотность 1,2506 кг/м³ (при н.у.).
В жидком состоянии (темп. кипения −195,8 °C) — бесцветная, подвижная, как вода, жидкость. Плотность жидкого азота 808 кг/м³. При контакте с воздухом поглощает из него кислород.
При −209,86 °C азот переходит в твердое состояние в виде снегоподобной массы или больших белоснежных кристаллов. При контакте с воздухом поглощает из него кислород, при этом плавится, образуя раствор кислорода в азоте.
Известны три кристаллические модификации твёрдого азота. В интервале 36,61 — 63,29 К существует фаза β-N2 с гексагональной плотной упаковкой, пространственная группа P63/mmc, параметры решётки a=3,93 Å и c=6,50 Å. При температуре ниже 36,61 К устойчива фаза α-N2 с кубической решёткой, имеющая пространственную группу Pa3 или P213 и период a=5,660 Å. Под давлением более 3500 атмосфер и температуре ниже 83 K образуется гексагональная фаза γ-N2.
Химические свойства, строение молекулы
Азот в свободном состоянии существует в форме двухатомных молекул N2, электронная конфигурация которых описывается формулой σs²σs*2πx, y4σz², что соответствует тройной связи между молекулами азота N≡N (длина связи dN≡N = 0,1095 нм). Вследствие этого молекула азота крайне прочна, для реакции диссоциации N2 ↔ 2N изменение энтальпии в реакции ΔH°298=945 кДж/моль[4], константа скорости реакции К298=10−120, то есть диссоциация молекул азота при нормальных условиях практически не происходит (равновесие практически полностью сдвинуто влево). Молекула азота неполярна и слабо поляризуется, силы взаимодействия между молекулами очень слабые, поэтому в обычных условиях азот газообразен.
Даже при 3000 °C степень термической диссоциации N2 составляет всего 0,1 %, и лишь при температуре около 5000 °C достигает нескольких процентов (при нормальном давлении). В высоких слоях атмосферы происходит фотохимическая диссоциация молекул N2. В лабораторных условиях можно получить атомарный азот, пропуская газообразный N2 при сильном разряжении через поле высокочастотного электрического разряда. Атомарный азот намного активнее молекулярного: в частности, при обычной температуре он реагирует с серой, фосфором, мышьяком и с рядом металлов, например, со ртутью.
Вследствие большой прочности молекулы азота некоторые его соединения эндотермичны (многие галогениды, азиды, оксиды), то есть энтальпия их образования положительна, а соединения азота термически малоустойчивы и довольно легко разлагаются при нагревании. Именно поэтому азот на Земле находится по большей части в свободном состоянии.
Ввиду своей значительной инертности азот при обычных условиях реагирует только с литием:

при нагревании он реагирует с некоторыми другими металлами и неметаллами, также образуя нитриды:


Наибольшее практическое значение имеет нитрид водорода (аммиак) NH3, получаемый взаимодействием водорода с азотом (см. ниже).
В электрическом разряде реагирует с кислородом, образуя оксид азота(II) NO.
Описано несколько десятков комплексов с молекулярным азотом.
Промышленное связывание атмосферного азота
Соединения азота чрезвычайно широко используются в химии, невозможно даже перечислить все области, где находят применение вещества, содержащие азот: это индустрия удобрений, взрывчатых веществ, красителей, медикаментов и проч. Хотя колоссальные количества азота доступны в прямом смысле слова «из воздуха», из-за описанной выше прочности молекулы азота N2 долгое время оставалась нерешённой задача получения соединений, содержащих азот, из воздуха; большая часть соединений азота добывалась из его минералов, таких, как чилийская селитра. Однако сокращение запасов этих полезных ископаемых, а также рост потребности в соединениях азота заставил форсировать работы по промышленному связыванию атмосферного азота.
Наиболее распространён аммиачный способ связывания атмосферного азота. Обратимая реакция синтеза аммиака:

экзотермическая (тепловой эффект 92 кДж) и идёт с уменьшением объёма, поэтому для сдвига равновесия вправо в соответствии с принципом Ле Шателье — Брауна необходимо охлаждение смеси и высокие давления. Однако с кинетической точки зрения снижение температуры невыгодно, так как при этом сильно снижается скорость реакции — уже при 700 °C скорость реакции слишком мала для её практического использования.
В таких случаях используется катализ, так как подходящий катализатор позволяет увеличить скорость реакции без сдвига равновесия. В процессе поиска подходящего катализатора было испробовано около двадцати тысяч различных соединений. По совокупности свойств (каталитическая активность, стойкость к отравлению, дешевизна) наибольшее применение получил катализатор на основе металлического железа с примесями оксидов алюминия и калия. Процесс ведут при температуре 400—600 °C и давлениях 10—1000 атмосфер.
Следует отметить, что при давлениях выше 2000 атмосфер синтез аммиака из смеси водорода и азота идёт с высокой скоростью и без катализатора. Например, при 850 °C и 4500 атмосфер выход продукта составляет 97 %.
Существует и ещё один, менее распространённый способ промышленного связывания атмосферного азота — цианамидный метод, основанный на реакции карбида кальция с азотом при 1000 °C. Реакция происходит по уравнению:

Реакция экзотермична, её тепловой эффект 293 кДж.
Ежегодно из атмосферы Земли промышленным путём отбирается примерно 1·106 т азота.
Соединения азота
Степени окисления азота в соединениях −3, −2, −1, 0, +1, +2, +3, +4, +5.
- Соединения азота в степени окисления −3 представлены нитридами, из которых практически наиболее важен аммиак;
- Соединения азота в степени окисления −2 менее характерны, представлены пернитридами, из которых самый важный пернитрид водорода N2H4 или гидразин (существует также крайне неустойчивый пернитрид водорода N2H2, диимид);
- Соединения азота в степени окисления −1 NH2OH (гидроксиламин) — неустойчивое основание, применяющееся, наряду с солями гидроксиламмония, в органическом синтезе;
- Соединения азота в степени окисления +1 оксид азота(I) N2O (закись азота, веселящий газ);
- Соединения азота в степени окисления +2 оксид азота(II) NO (монооксид азота);
- Соединения азота в степени окисления +3 оксид азота(III) N2O3, азотистая кислота, производные аниона NO2−, трифторид азота (NF3);
- Соединения азота в степени окисления +4 оксид азота(IV) NO2 (диоксид азота, бурый газ);
- Соединения азота в степени окисления +5 оксид азота(V) N2O5, азотная кислота, её соли — нитраты и другие производные, а также тетрафтораммоний NF4+ и его соли.
Применение

![]()
Слабокипящий жидкий азот в металлическом стакане.
Жидкий азот применяется как хладагент и для криотерапии.
Промышленные применения газообразного азота обусловлены его инертными свойствами. Газообразный азот пожаро- и взрывобезопасен, препятствует окислению, гниению. В нефтехимии азот применяется для продувки резервуаров и трубопроводов, проверки работы трубопроводов под давлением, увеличения выработки месторождений. В горнодобывающем деле азот может использоваться для создания в шахтах взрывобезопасной среды, для распирания пластов породы. В производстве электроники азот применяется для продувки областей, не допускающих наличия окисляющего кислорода. Если в процессе, традиционно проходящем с использованием воздуха, окисление или гниение являются негативными факторами — азот может успешно заместить воздух.
Важной областью применения азота является его использование для дальнейшего синтеза самых разнообразных соединений, содержащих азот, таких, как аммиак, азотные удобрения, взрывчатые вещества, красители и т. п. Большие количества азота используются в коксовом производстве («сухое тушение кокса») при выгрузке кокса из коксовых батарей, а также для «передавливания» топлива в ракетах из баков в насосы или двигатели.
В пищевой промышленности азот зарегистрирован в качестве пищевой добавки E941, как газовая среда для упаковки и хранения, хладагент, а жидкий азот применяется при разливе масел и негазированных напитков для создания избыточного давления и инертной среды в мягкой таре.
Газообразным азотом заполняют камеры шин шасси летательных аппаратов. Кроме того, в последнее время заполнение шин азотом стало популярно и среди автолюбителей, хотя однозначных доказательств эффективности использования азота вместо воздуха для наполнения автомобильных шин нет.
Жидкий азот нередко демонстрируется в кинофильмах в качестве вещества, способного мгновенно заморозить достаточно крупные объекты. Это широко распространённое заблуждение. Даже для замораживания цветка необходимо достаточно продолжительное время. Это связано отчасти с весьма низкой теплоёмкостью азота. По этой же причине весьма затруднительно охлаждать, скажем, замки до −196 °C и раскалывать их одним ударом.
Литр жидкого азота, испаряясь и нагреваясь до 20 °C, образует примерно 700 литров газа. По этой причине жидкий азот хранят в специальных сосудах Дьюара с вакуумной изоляцией открытого типа или криогенных ёмкостях под давлением. На этом же факте основан принцип тушения пожаров жидким азотом. Испаряясь, азот вытесняет кислород, необходимый для горения, и пожар прекращается. Так как азот, в отличие от воды, пены или порошка, просто испаряется и выветривается, азотное пожаротушение — самый эффективный с точки зрения сохранности ценностей механизм тушения пожаров.
Заморозка жидким азотом живых существ с возможностью последующей их разморозки проблематична. Проблема заключается в невозможности заморозить (и разморозить) существо достаточно быстро, чтобы неоднородность заморозки не сказалась на его жизненных функциях. Станислав Лем, фантазируя на эту тему в книге «Фиаско», придумал экстренную систему заморозки азотом, в которой шланг с азотом, выбивая зубы, вонзался в рот астронавта и внутрь его подавался обильный поток азота.
Маркировка баллонов
Баллоны с азотом окрашены в чёрный цвет, должны иметь надпись жёлтого цвета и коричневую полосу (согласно нормам РФ).
Интересные факты
Цитата из Большой Советской Энциклопедии издания 1952 г. (том 1, стр. 452, статья «Азот»):
Азот в сложении с капитализмом — это война, разрушение, смерть. Азот в сложении с социализмом — это высокий урожай, высокая производительность труда, высокий материальный и культурный уровень трудящихся.
См. также
- Категория:Соединения азота;
- Оксиды азота;
- Аммиак;
- Азотная кислота;
- Гидразин;
- Гидроксид аммония;
- Селитры;
- Нитраты.
- Азотистый обмен почвы
- Криогенная резка
- Азотное правило
- Азотная станция
Примечания
- ↑ Nitrogen: electronegativities (англ.). WebElements. Проверено 5 августа 2010.
- ↑ Кнунянц И. Л. (гл. ред.) Химическая энциклопедия: в 5 тт. — Москва: Советская энциклопедия, 1988. — Т. 1. — С. 58. — 623 с. — 100 000 экз.
- ↑ Фигуровский Н. А. Открытие элементов и происхождение их названий. — М.: Наука, 1970. 207 с.
- ↑ Nitrogen atom
Литература
- Некрасов Б. В., Основы общей химии, т. 1, М.: «Химия», 1973;
- Химия: Справ. изд./В. Шретер, К.-Х. Лаутеншлегер, Х. Бибрак и др.: Пер. с нем. 2-е изд., стереотип. — М.: Химия, 2000 ISBN 5-7245-0360-3 (рус.), ISBN 3-343-00208-9 (нем.);
- Ахметов Н. С., Общая и неорганическая химия. 5-е изд., испр. — М.: Высшая школа, 2003 ISBN 5-06-003363-5;
- Гусакова Н. В., Химия окружающей среды. Серия «Высшее образование». Ростов-на-Дону: Феникс, 2004 ISBN 5-222-05386-5;
- Исидоров В. А., Экологическая химия. СПб: Химиздат, 2001 ISBN 5-7245-1068-5;
- Трифонов Д. Н., Трифонов В. Д., Как были открыты химические элементы — М.: Просвещение, 1980
- Справочник химика, 2-е изд., т. 1, М.: «Химия», 1966;
Ссылки
| Азот на Викискладе? |
- Азот, химический элемент // Энциклопедический словарь Брокгауза и Ефрона: В 86 томах (82 т. и 4 доп.). — СПб., 1890—1907.
- Азот на Webelements
- Азот в Популярной библиотеке химических элементов
| Периодическая система химических элементов Д. И. Менделеева | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | |||||||||||||||
| 1 | H | He | ||||||||||||||||||||||||||||||
| 2 | Li | Be | B | C | N | O | F | Ne | ||||||||||||||||||||||||
| 3 | Na | Mg | Al | Si | P | S | Cl | Ar | ||||||||||||||||||||||||
| 4 | K | Ca | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | Ga | Ge | As | Se | Br | Kr | ||||||||||||||
| 5 | Rb | Sr | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd | In | Sn | Sb | Te | I | Xe | ||||||||||||||
| 6 | Cs | Ba | La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg | Tl | Pb | Bi | Po | At | Rn |
| 7 | Fr | Ra | Ac | Th | Pa | U | Np | Pu | Am | Cm | Bk | Cf | Es | Fm | Md | No | Lr | Rf | Db | Sg | Bh | Hs | Mt | Ds | Rg | Cn | Uut | Fl | Uup | Lv | Uus | Uuo |
|
Азот

4.4
Средняя оценка: 4.4
Всего получено оценок: 626.
4.4
Средняя оценка: 4.4
Всего получено оценок: 626.
Азот экспериментальным путем был обнаружен шотландским химиком Д. Резерфордом в 1772 году. В природе азот находится в основном в свободном состоянии и является одной из главных составляющих воздуха. Каковы же физические и химические свойства азота?
Общая характеристика
Азот – химический элемент V группы периодической системы Менделеева, атомный номер 7, атомная масса 14, формула азота – N2. Перевод названия элемента – «безжизненный» – может относится к азоту как к простому веществу. Однако азот в связанном состоянии является одним из главных элементов жизни, входит в состав белков, нуклеиновых кислот, витаминов и т.д.

Азот – элемент второго периода, не имеет возбужденных состояний, так как атом не имеет свободных орбиталей. Но этот химический элемент может проявлять в основном состоянии валентность не только III, но и IV за счет образования ковалентной связи по донорно-акцепторному механизму с участием неподеленной электронной пары азота. Степень окисления, которую может проявлять азот, изменяется в широких пределах от -3 до +5.
при изучении строения молекулы азота необходимо помнить, что химическая связь осуществляется за счет трех общих пар p-электронов, орбитали которых направлены по осям x, y, z.
Химические свойства азота
В природе азот встречается в виде простого вещества – газа N2 (объемная доля в воздухе 78%) и в связанном состоянии. В молекуле азота атомы связаны прочной тройной связью. Энергия этой связи составляет 940 кДж/моль. При обычной температуре азот может взаимодействовать только с литием (Li3 N). После предварительной активизации молекул путем нагревания, облучения или действием катализаторов азот вступает в реакции с металлами и неметаллами. Азот может вступать в реакции с магнием, кальцием или, например, алюминием:
3Mg+N2=Mg3N2
3Ca+N2=Ca3N2
2Al+N2=2AlN
Особенно важен синтез аммиака из простых веществ – азота и водорода в присутствии катализатора (губчатое железо):N2+3H2=2NH3+Q. Аммиак – бесцветный газ с резким запахом. Он хорошо растворим в воде, что в значительной степени обусловлено образованием водородных связей между молекулами аммиака и воды, а также реакцией присоединения к воде по донорно-акцепторному механизму. Слабощелочная реакция раствора обусловлена наличием в растворе ионов OH- (в небольшой концентрации, так как степень диссоциации гидроксида аммония очень мала – это слабое растворимое основание).

Из шести оксидов азота – N2O, NO, N2O3, NO2, N2O4, N2O5, где азот проявляет степень окисления от +1 до +5, два первых – N2O и NO – несолеобразующие, остальные вступают в реакцию с образованием солей.
Азотную кислоту, самое важное соединение азота, в промышленности получают из аммиака в 3 стадии:
- окисление аммиака на платиновом катализаторе:
4NH3 +5O2=4NO+6H2O
- окисление NO до NO2 кислородом воздуха:
2NO+O2=2NO2
- поглощение NO2 водой в избытке кислорода:
4NO2 +2H2O+O2=4HNO3
Азот также может реагировать при высоких температурах и давлении (в присутствии катализатора) с водородом:
N2+3H2=2NH3

Применение азота
Основное применение азот находит в качестве исходного продукта для синтеза аммиака, а также для производства азотной кислоты, минеральных удобрений, красителей, взрывчатых веществ и других азотосодержащих соединений. Жидкий азот используют в охладительных системах. Для придания стали большей твердости, увеличения износостойкости, коррозионной стойкости и теплостойкости ее поверхность насыщают азотом при высоких температурах. Такая сталь выдерживает нагревание до 500 градусов без потери своей твердости.

Что мы узнали?
Азот – бесцветный газ без запаха, цвета и вкуса. В школьном курсе химии изучаются его основные свойства, а также способность вступать в реакцию с металлами и неметаллами при нормальных условиях, а также под действием катализаторов.
Тест по теме
Доска почёта
Чтобы попасть сюда — пройдите тест.
-

Фарида Гаскарова
10/10
-

Александр’ Шишиков
8/10
-

Сергей Ефремов
8/10
-

Андрей Борисов
9/10
Оценка доклада
4.4
Средняя оценка: 4.4
Всего получено оценок: 626.
А какая ваша оценка?
| Азот | |
|---|---|
| Атомный номер | 7 |
| Внешний вид простого вещества | Газ без цвета вкуса и запаха, химически весьма инертен |
| Свойства атома | |
| Атомная масса (молярная масса) | 14,00674 а. е. м. (г/моль) |
| Радиус атома | 92 пм |
| Энергия ионизации (первый электрон) | 1401,5 (14,53) кДж/моль (эВ) |
| Электронная конфигурация | [He] 2s2 2p3 |
| Химические свойства | |
| Ковалентный радиус | 75 пм |
| Радиус иона | 13 (+5e) 171 (-3e) пм |
| Электроотрицательность (по Полингу) | 3,04 |
| Электродный потенциал | — |
| Степени окисления | 5, 4, 3, 2, 1, 0, -1, -3 |
| Термодинамические свойства простого вещества | |
| Плотность | 0,808 (−195,8 °C) г/см³ |
| Молярная теплоёмкость | 29,125 (газ N2) Дж/(K·моль) |
| Теплопроводность | 0,026 Вт/(м·K) |
| Температура плавления | 63,29 K |
| Теплота плавления | (N2) 0.720 кДж/моль |
| Температура кипения | 77,4 K |
| Теплота испарения | (N2) 5.57 кДж/моль |
| Молярный объём | 17,3 см³/моль |
| Кристаллическая решётка простого вещества | |
| Структура решётки | кубическая |
| Параметры решётки | 5,661 Å |
| Отношение c/a | — |
| Температура Дебая | n/a K |
| N | 7 |
| 14,00674 | |
| [He]2s22p3 | |
| Азот |
Азот — элемент главной подгруппы пятой группы второго периода периодической системы химических элементов Д. И. Менделеева с атомным номером 7. Обозначается символом N (Nitrogenium). Простое вещество азот (CAS-номер: 7727-37-9) — достаточно инертный при нормальных условиях двухатомный газ без цвета, вкуса и запаха (формула N2).
Открытие
В 1777 году Генри Кавендиш провёл следующий опыт: он многократно пропускал воздух над раскалённым углём, затем обрабатывал его щёлочью, в результате получался остаток, который Кавендиш назвал удушливым (или мефитическим) воздухом. С позиций современной химии ясно, что в реакции с раскалённым углём кислород воздуха связывался в углекислый газ, который затем реагировал со щёлочью. При этом остаток газа представлял собой по большей части азот. Таким образом, Кавендиш выделил азот, но не сумел понять, что это новое простое вещество (химический элемент). В том же году Кавендиш сообщил об этом опыте Джозефу Пристли.
Пристли в это время проводил серию экспериментов, в которых также связывал кислород воздуха и удалял полученный углекислый газ, то есть также получал азот, однако, будучи сторонником господствующей в те времена теории флогистона, совершенно неверно истолковал полученные результаты (по его мнению, процесс был противоположным — не кислород удалялся из газовой смеси, а наоборот, в результате обжига воздух насыщался флогистоном; оставшийся воздух (азот) он и назвал насыщенным флогистоном, то есть флогистированным). Очевидно, что и Пристли, хотя и смог выделить азот, не сумел понять сути своего открытия, поэтому и не считается первооткрывателем азота.
Одновременно схожие эксперименты с тем же результатом проводил и Карл Шееле.
В 1772 году азот (под названием «испорченного воздуха») как простое вещество описал Даниэль Резерфорд, он опубликовал магистерскую диссертацию, где указал основные свойства азота (не реагирует со щелочами, не поддерживает горения, непригоден для дыхания). Именно Даниэль Резерфорд и считается первооткрывателем азота.
В дальнейшем азот был изучен Генри Кавендишем (интересен тот факт, что он сумел связать азот с кислородом при помощи разрядов электрического тока, а после поглощения оксидов азота в остатке получил небольшое количество газа, абсолютно инертного, хотя, как и в случае с азотом, не смог понять, что выделил новые химические элементы — инертные газы). Однако и Резерфорд был сторонником флогистонной теории, поэтому также не смог понять, что же он выделил. Таким образом, чётко определить первооткрывателя азота невозможно.
Происхождение названия
Азот ( ἀζωτος — безжизненный, Nitrogenium), вместо предыдущих названий («флогистированный», «мефитический» и «испорченный» воздух) предложил в 1787 году Антуан Лавуазье, который в то время в составе группы других французских учёных разрабатывал принципы химической номенклатуры. Как показано выше, в то время уже было известно, что азот не поддерживает ни горения, ни дыхания. Это свойство и сочли наиболее важным. Хотя впоследствии выяснилось, что азот, наоборот, крайне необходим для всех живых существ, название сохранилось во французском и русском языках.
Существует и иная версия. Слово «азот» придумано не Лавуазье и не его коллегами по номенклатурной комиссии; оно вошло в алхимическую литературу уже в раннем средневековье и употреблялось для обозначения «первичной материи металлов», которую считали «альфой и омегой» всего сущего. Это выражение заимствовано из Апокалипсиса: «Я есмь Альфа и Омега, начало и конец». Слово составлено из начальных и конечных букв алфавитов трёх языков — латинского, греческого и древнееврейского, — считавшихся «священными», поскольку, согласно Евангелиям, надпись на кресте при распятии Христа была сделана на этих языках (а, альфа, алеф и зет, омега, тав — AAAZOTH). Составители новой химической номенклатуры хорошо знали о существовании этого слова; инициатор её создания Гитон де Морво отмечал в своей «Методической энциклопедии» (1786) алхимическое значение термина.
На латыни азот называется «Nitrogenium», то есть «рождающий селитру»; английское название производится от латинского. В немецком языке используется название Stickstoff, что означает «удушающее вещество».
Азот в природе
Изотопы азоты
Природный азот состоит из двух стабильных изотопов 14N — 99,635 % и 15N — 0,365 %.
Известны радиоактивные изотопы азота с массовыми числами 11, 12, 13, 16 и 17. Все они являются очень короткоживущими изотопами. Самый стабильный из них 13N имеет период полураспада 10 мин.
Магнитный момент ядер изотопов 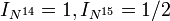
Распространённость
Вне пределов Земли азот обнаружен в газовых туманностях, солнечной атмосфере, на Уране, Нептуне, межзвёздном пространстве и др. Азот — четвёртый по распространённости элемент Солнечной системы (после водорода, гелия и кислорода).
Азот, в форме двухатомных молекул N2 составляет большую часть атмосферы, где его содержание составляет 75,6 % (по массе) или 78,084 % (по объёму), то есть около 3,87·1015 т.
Содержание азота в земной коре, по данным разных авторов, составляет (0,7—1,5)·1015 т (причём в гумусе — порядка 6·1010 т), а в мантии Земли — 1,3·1016 т. Такое соотношение масс заставляет предположить, что главным источником азота служит верхняя часть мантии, откуда он поступает в другие оболочки Земли с извержениями вулканов.
Масса растворённого в гидросфере азота, учитывая, что одновременно происходят процессы растворения азота атмосферы в воде и выделения его в атмосферу, составляет около 2·1013 т, кроме того примерно 7·1011 т азота содержатся в гидросфере в виде соединений.
Биологическая роль
Азот является элементом, необходимым для существования животных и растений, он входит в состав белков (16—18 % по массе), аминокислот, нуклеиновых кислот, нуклеопротеидов, хлорофилла, гемоглобина и др. В составе живых клеток по числу атомов азота около 2%, по массовой доле — около 2,5 % (четвертое место после водорода, углерода и кислорода). В связи с этим значительное количество связанного азота содержится в живых организмах, «мёртвой органике» и дисперсном веществе морей и океанов. Это количество оценивается примерно в 1,9·1011 т. В результате процессов гниения и разложения азотсодержащей органики, при условии благоприятных факторов окружающей среды, могут образоваться природные залежи полезных ископаемых, содержащие азот, например, «чилийская селитра» (нитрат натрия с примесями других соединений), норвежская, индийская селитры.
Круговорот азота в природе
-
Круговорот азота в природе
Фиксация атмосферного азота в природе происходит по двум основным направлениям — абиогенному и биогенному. Первый путь включает главным образом реакции азота с кислородом. Так как азот химически весьма инертен, для окисления требуются большие количества энергии (высокие температуры). Эти условия достигаются при разрядах молний, когда температура достигает 25000 °C и более. При этом происходит образование различных оксидов азота. Существует также вероятность, что абиотическая фиксация происходит в результате фотокаталитических реакций на поверхности полупроводников или широкополосных диэлектриков (песок пустынь).
Однако основная часть молекулярного азота (около 1,4·108 т/год) фиксируется биотическим путём. Долгое время считалось, что связывать молекулярный азот могут только небольшое количество видов микроорганизмов (хотя и широко распространённых на поверхности Земли): бактерии Azotobacter и Clostridium, клубеньковые бактерии бобовых растений Rhizobium, цианобактерии Anabaena, Nostoc и др. Сейчас известно, что этой способностью обладают многие другие организмы в воде и почве, например, актиномицеты в клубнях ольхи и других деревьев (всего 160 видов). Все они превращают молекулярный азот в соединения аммония (NH4+). Этот процесс требует значительных затрат энергии (для фиксации 1 г атмосферного азота бактерии в клубеньках бобовых расходуют порядка 167,5 кДж, то есть окисляют примерно 10 г глюкозы). Таким образом, видна взаимная польза от симбиоза растений и азотфиксирующих бактерий — первые предоставляют вторым «место для проживания» и снабжают полученным в результате фотосинтеза «топливом» — глюкозой, вторые обеспечивают необходимый растениям азот в усваиваемой ими форме.
Азот в форме аммиака и соединений аммония, получающийся в процессах биогенной азотфиксации, быстро окисляется до нитратов и нитритов (этот процесс носит название нитрификации). Последние, не связанные тканями растений (и далее по пищевой цепи травоядными и хищниками), недолго остаются в почве. Большинство нитратов и нитритов хорошо растворимы, поэтому они смываются водой и в конце концов попадают в мировой океан (этот поток оценивается в 2,5—8·107 т/год).
Азот, включённый в ткани растений и животных, после их гибели подвергается аммонификации (разложению содержащих азот сложных соединений с выделением аммиака и ионов аммония) и денитрификации то есть выделению атомарного азота, а также его оксидов. Эти процессы целиком происходят благодаря деятельности микроорганизмов в аэробных и анаэробных условиях.
В отсутствие деятельности человека процессы связывания азота и нитрификации практически полностью уравновешены противоположными реакциями денитрификации. Часть азота поступает в атмосферу из мантии с извержениями вулканов, часть прочно фиксируется в почвах и глинистых минералах, кроме того, постоянно идёт утечка азота из верхних слоёв атмосферы в межпланетное пространство.
Токсикология азота и его соединений
Сам по себе атмосферный азот достаточно инертен, чтобы оказывать непосредственное влияние на организм человека и млекопитающих. Тем не менее, при повышенном давлении он вызывает наркоз, опьянение или удушье (при недостатке кислорода); при быстром снижении давления азот вызывает кессонную болезнь.
Многие соединения азота очень активны и нередко токсичны.
Получение
В лабораториях его можно получать по реакции разложения нитрита аммония:
NH4NO2 → N2↑ + 2H2O
Реакция экзотермическая, идёт с выделением 80 ккал (335 кДж), поэтому требуется охлаждение сосуда при её протекании (хотя для начала реакции требуется нагревание нитрита аммония).
Практически эту реакцию выполняют, добавляя по каплям насыщенный раствор нитрита натрия в нагретый насыщенный раствор сульфата аммония, при этом образующийся в результате обменной реакции нитрит аммония мгновенно разлагается.
Выделяющийся при этом газ загрязнён аммиаком, оксидом азота (I) и кислородом, от которых его очищают, последовательно пропуская через растворы серной кислоты, сульфата железа (II) и над раскалённой медью. Затем азот осушают.
Ещё один лабораторный способ получения азота — нагревание смеси дихромата калия и сульфата аммония (в соотношении 2:1 по массе). Реакция идёт по уравнениям:
K2Cr2O7 + (NH4)2SO4 = (NH4)2Cr2O7 + K2SO4
(NH4)2Cr2O7 →(t) Cr2O3 + N2↑ + 4H2O
Самый чистый азот можно получить разложением азидов металлов:
2NaN3 →(t) 2Na + 3N2↑
Так называемый «воздушный», или «атмосферный» азот, то есть смесь азота с благородными газами, получают путём реакции воздуха с раскалённым коксом:
O2+ 4N2 + 2C → 2CO + 4N2
При этом получается так называемый «генераторный», или «воздушный», газ — сырьё для химических синтезов и топливо. При необходимости из него можно выделить азот, поглотив монооксид углерода.
Молекулярный азот в промышленности получают фракционной перегонкой жидкого воздуха. Этим методом можно получить и «атмосферный азот». Также широко применяются азотные установки, в которых используется метод адсорбционного и мембранного газоразделения.
Один из лабораторных способов — пропускание аммиака над оксидом меди (II) при температуре ~700°С:
2NH3 + 3CuO → N2↑ + 3H2O + 3Cu
Аммиак берут из его насыщенного раствора при нагревании. Количество CuO в 2 раза больше расчётного. Непосредственно перед применением азот очищают от примеси кислорода и аммиака пропусканием над медью и её оксидом (II) (тоже ~700°C), затем сушат концентрированной серной кислотой и сухой щёлочью. Процесс происходит довольно медленно, но он того стоит: газ получается весьма чистый.
Свойства
Физические свойства
![]()
![]()
Оптический линейчатый эмиссионный спектр азота

![]()
Схема электронных оболочек атома азота
При нормальных условиях азот это бесцветный газ, не имеет запаха, мало растворим в воде (2,3 мл/100г при 0 °C, 0,8 мл/100г при 80 °C).
В жидком состоянии (темп. кипения -195,8 °C) – бесцветная, подвижная, как вода, жидкость. При контакте с воздухом поглощает из него кислород.
При -209,86 °C азот переходит в твердое состояние в виде снегоподобной массы или больших белоснежных кристаллов. При контакте с воздухом поглощает из него кислород, при этом плавится, образуя раствор кислорода в азоте.
Известны три кристаллические модификации твёрдого азота. В интервале 36,61 – 63,29 К существует фаза β-N2 с гексагональной плотной упаковкой, пространственная группа P63/mmc, параметры решётки a=3,93 Å и c=6,50 Å. При температуре ниже 36,61 К устойчива фаза α-N2 с кубической решёткой, имеющая пространственную группу Pa3 или P213 и период a=5,660 Å. Под давлением более 3500 атмосфер и температуре ниже 83 K образуется гексагональная фаза γ-N2.
Химические свойства, строение молекулы
Азот в свободном состоянии существует в форме двухатомных молекул N2, электронная конфигурация которых описывается формулой σs²σs*2πx, y4σz², что соответствует тройной связи между молекулами азота N≡N (длина связи dN≡N = 0,1095 нм). Вследствие этого молекула азота крайне прочна, для реакции диссоциации N2 ↔ 2N удельная энтальпия образования ΔH°298=945 кДж, константа скорости реакции К298=10-120, то есть диссоциация молекул азота при нормальных условиях практически не происходит (равновесие практически полностью сдвинуто влево). Молекула азота неполярна и слабо поляризуется, силы взаимодействия между молекулами очень слабые, поэтому в обычных условиях азот газообразен.
Даже при 3000 °C степень термической диссоциации N2 составляет всего 0,1 %, и лишь при температуре около 5000 °C достигает нескольких процентов (при нормальном давлении). В высоких слоях атмосферы происходит фотохимическая диссоциация молекул N2. В лабораторных условиях можно получить атомарный азот, пропуская газообразный N2 при сильном разряжении через поле высокочастотного электрического разряда. Атомарный азот намного активнее молекулярного: в частности, при обычной температуре он реагирует с серой, фосфором, мышьяком и с рядом металлов, например, со ртутью.
Вследствие большой прочности молекулы азота многие его соединения эндотермичны, энтальпия их образования отрицательна, а соединения азота термически малоустойчивы и довольно легко разлагаются при нагревании. Именно поэтому азот на Земле находится по большей части в свободном состоянии.
Ввиду своей значительной инертности азот при обычных условиях реагирует только с литием:
6Li + N2 → 2Li3N,
при нагревании он реагирует с некоторыми другими металлами и неметаллами, также образуя нитриды:
3Mg + N2 → Mg3N2,
2B + N2 →2BN,
Наибольшее практическое значение имеет нитрид водорода (аммиак):
Промышленное связывание атмосферного азота
Соединения азота чрезвычайно широко используются в химии, невозможно даже перечислить все области, где находят применение вещества, содержащие азот: это индустрия удобрений, взрывчатых веществ, красителей, медикаментов и проч. Хотя колоссальные количества азота доступны в прямом смысле слова «из воздуха», из-за описанной выше прочности молекулы азота N2 долгое время оставалась нерешённой задача получения соединений, содержащих азот, из воздуха; большая часть соединений азота добывалась из его минералов, таких, как чилийская селитра. Однако сокращение запасов этих полезных ископаемых, а также рост потребности в соединениях азота заставил форсировать работы по промышленному связыванию атмосферного азота.
Наиболее распространён аммиачный способ связывания атмосферного азота. Обратимая реакция синтеза аммиака:
3H2 + N2 ↔ 2NH3
экзотермическая (тепловой эффект 92 кДж) и идёт с уменьшением объёма, поэтому для сдвига равновесия вправо в соответствии с принципом Ле Шателье — Брауна необходимо охлаждение смеси и высокие давления. Однако с кинетической точки зрения снижение температуры невыгодно, так как при этом сильно снижается скорость реакции — уже при 700 °C скорость реакции слишком мала для её практического использования.
В таких случаях используется катализ, так как подходящий катализатор позволяет увеличить скорость реакции без сдвига равновесия. В процессе поиска подходящего катализатора было испробовано около двадцати тысяч различных соединений. По совокупности свойств (каталитическая активность, стойкость к отравлению, дешевизна) наибольшее применение получил катализатор на основе металлического железа с примесями оксидов алюминия и калия. Процесс ведут при температуре 400—600°С и давлениях 10—1000 атмосфер.
Следует отметить, что при давлениях выше 2000 атмосфер синтез аммиака из смеси водорода и азота идёт с высокой скоростью и без катализатора. Например, при 850 °C и 4500 атмосфер выход продукта составляет 97 %.
Существует и ещё один, менее распространённый способ промышленного связывания атмосферного азота — цианамидный метод, основанный на реакции карбида кальция с азотом при 1000 °C. Реакция происходит по уравнению:
CaC2 + N2 → CaCN2 + C.
Реакция экзотермична, её тепловой эффект 293 кДж.
Ежегодно из атмосферы Земли промышленным путём отбирается примерно 1·106 т азота. Подробно процесс получения азота изложен здесь ГРАСИС
Соединения азота
- Степени окисления азота в соединениях −3, −2, −1, +1, +2, +3, +4, +5.
Соединения азота в степени окисления −3 представлены нитридами, из которых практически наиболее важен аммиак;
Соединения азота в степени окисления −2 менее характерны, представлены пернитридами, из которых самый важный пернитрид водорода N2H4 или гидразин (существует также крайне неустойчивый пернитрид водорода N2H2, диимид);
Соединения азота в степени окисления −1 NH2OH (гидроксиламин) — неустойчивое основание, применяющееся, наряду с солями гидроксиламмония, в органическом синтезе;
Соединения азота в степени окисления +1 оксид азота (I) N2O (закись азота, веселящий газ);
Соединения азота в степени окисления +2 оксид азота (II) NO (монооксид азота);
Соединения азота в степени окисления +3 оксид азота (III) N2O3, азотистая кислота, производные аниона NO2-, трифторид азота NF3;
Соединения азота в степени окисления +4 оксид азота (IV) NO2 (диоксид азота, бурый газ);
Соединения азота в степени окисления +5 — оксид азота (V) N2O5, азотная кислота и её соли — нитраты, и др.
Использование и применение

![]()
Слабокипящий жидкий азот в металлическом стакане.
Жидкий азот применяется как хладагент и для криотерапии.
Промышленные применения газообразного азота обусловлены его инертными свойствами. Газообразный азот пожаро- и взрывобезопасен, препятствует окислению, гниению. В нефтехимии азот применяется для продувки резервуаров и трубопроводов, проверки работы трубопроводов под давлением, увеличения выработки месторождений. В горнодобывающем деле азот может использоваться для создания в шахтах взрывобезопасной среды, для распирания пластов породы. В производстве электроники азот применяется для продувки областей, не допускающих наличия окисляющего кислорода. Если в процессе, традиционно проходящем с использованием воздуха, окисление или гниение являются негативными факторами — азот может успешно заместить воздух.
Важной областью применения азота является его использование для дальнейшего синтеза самых разнообразных соединений, содержащих азот, таких, как аммиак, азотные удобрения, взрывчатые вещества, красители и т. п. Большие количества азота используются в коксовом производстве («сухое тушение кокса») при выгрузке кокса из коксовых батарей, а также для «передавливания» топлива в ракетах из баков в насосы или двигатели.
В пищевой промышленности азот зарегистрирован в качестве пищевой добавки E941, как газовая среда для упаковки и хранения, хладагент, а жидкий азот применяется при розливе масел и негазированных напитков для создания избыточного давления и инертной среды в мягкой таре.
Жидкий азот нередко демонстрируется в кинофильмах в качестве вещества, способного мгновенно заморозить достаточно крупные объекты. Это широко распространённая ошибка. Даже для замораживания цветка необходимо достаточно продолжительное время. Это связано отчасти с весьма низкой теплоёмкостью азота. По этой же причине весьма затруднительно охлаждать, скажем, замки до −196 °C и раскалывать их одним ударом.
Литр жидкого азота, испаряясь и нагреваясь до 20 °C, образует примерно 700 литров газа. По этой причине жидкий азот хранят в специальных сосудах Дьюара с вакуумной изоляцией открытого типа или криогенных ёмкостях под давлением. На этом же факте основан принцип тушения пожаров жидким азотом. Испаряясь, азот вытесняет кислород, необходимый для горения, и пожар прекращается. Так как азот, в отличие от воды, пены или порошка, просто испаряется и выветривается, азотное пожаротушение — самый эффективный с точки зрения сохранности ценностей механизм тушения пожаров.
Заморозка жидким азотом живых существ с возможностью последующей их разморозки проблематична. Проблема заключается в невозможности заморозить (и разморозить) существо достаточно быстро, чтобы неоднородность заморозки не сказалась на его жизненных функциях. Станислав Лем, фантазируя на эту тему в книге «Фиаско», придумал экстренную систему заморозки азотом, в которой шланг с азотом, выбивая зубы, вонзался в рот астронавта и внутрь его подавался обильный поток азота.
Маркировка баллонов
Баллоны с азотом окрашены в чёрный цвет, должны иметь надпись жёлтого цвета и коричневую полосу(нормыРоссийской Федерации).
Дополнительные материалы:
Соединения азота
Оксиды азота
Аммиак
Азотная кислота
Гидразин
Гидроксид аммония
Селитры
Нитраты
Азотистый обмен почвы
Криогенная резка
Азот, Nitrogenium, N (7)
Азот (Nitrogen, франц. Azote, нем. Stickstoff) был открыт почти одновременно несколькими исследователями. Кавендиш получил азот из воздуха (1772), пропуская последний через раскаленный уголь, а затем через раствор щелочи для поглощения углекислоты. Кавендиш не дал специального названия новому газу, упоминая о нем как о мефитическом воздухе (Air mephitic от латинского mephitis — удушливое или вредное испарение земли). Вскоре Пристлей установил, что если в воздухе долгое время горит свеча или находится животное (мышь), то такой воздух становится непригодным для дыхания. Официально открытие азота обычно приписывается ученику Блэка — Рутерфорду, опубликовавшему в 1772 г. диссертацию (на степень доктора медицины), — «О фиксируемом воздухе, называемом иначе удушливым», где впервые описаны некоторые химические свойства азота. В ати же годы Шееле получил азот из атмосферного воздуха тем же путем, что и Кавендиш. Он назвал новый ‘газ испорченным воздухом (Verdorbene Luft). Поскольку пропускание воздуха через раскаленный уголь рассматривалось химиками-флогистиками как его флогистирование, Пристлей (1775) назвал азот флогистированным воздухом — Air phlogisticated. О флогистировании воздуха в своем опыте говорил ранее и Кавендиш. Лавуазье в 1776 — 1777 гг. подробно исследовал состав атмосферного воздуха и установил, что 4/5 его объема состоят из удушливого газа (Аir mofette — атмосферный мофетт, или просто Mofett). Названия азота — флогистированный воздух, мефитический воздух, атмосферный мофетт, испорченный воздух и некоторые другие — употреблялись до признания в европейских странах новой химической номенклатуры, т. е. до выхода в свет известной книги «Метод химической номенклатуры» (1787).
Составители этой книги — члены номенклатурной комиссии Парижской академии наук — Гитон де Морво, Лавуазье, Бертолле и Фуркруа — приняли лишь несколько новых названий простых веществ, в частности, предложенные Лавуазье названия «кислород» и «водород». При выборе нового названия для азота комиссия, исходившая из принципов кислородной теории, оказалась в затруднении. Как известно, Лавуазье предлагал давать простым веществам такие названия, которые отражали бы их основнйе химические свойства. Соответственно этому азоту следовало бы дать название «радикал нитрик» или «радикал селитряной кислоты». Такие названия, пишет Лавуазье в своей книге «Начала элементарной химии» (1789), основаны на старых терминах нитр или селитра, принятых в искусствах, в химии и в обществе. Они были бы весьма подходящими, но известно, что азот является также основанием летучей щелочи (аммиака), как это было незадолго до этого установлено Бертолле. Поэтому название радикал, или основание селитряной кислоты, не отражает основных химических свойств азота. Не лучше ли остановиться на слове азот, которое, по мнению членов номенклатурной комиссии, отражает основное свойство элемента — его непригодность для дыхания и жизни. Авторы химической номенклатуры предложили производить слово азот от греческой отрицательной приставки «а» и слова жизнь. Таким образом, название азот, по их мнению, отражало его нежизненность, или безжизненность.
Однако слово азот придумано не Лавуазье и не его коллегами по комиссии. Оно известно с древности и употреблялось философами и алхимиками средневековья для обозначения «первичной материи (основы) металлов», так называемого меркурия философов, или двойного меркурия алхимиков. Слово азот вошло в литературу, вероятно, в первые столетия средневековья, как и многие другие зашифрованные и имевшие мистический смысл названия. Оно встречается в сочинениях многих алхимиков, начиная с Бэкона (ХIII в.),- у Парацельса, Либавия, Валентина и др. Либавий указывает даже, что слово азот (azoth) происходит от старинного испано-арабского слова азок (azoque или azoc), обозначавшего ртуть. Но более вероятно, что эти слова появились в результате искажений переписчиками коренного слова азот (azot или azoth). Теперь происхождение слова азот установлено более точно. Древние философы и алхимики считали «первичную материю металлов» альфой и омегой всего существующего. В свою очередь это выражение заимствовано из Апокалипсиса — последней книги Библии: «я — альфа и омега, начало и конец, первый и последний». В древности и в средние века христианские философы считали приличным употреблять при написании своих трактатов только три языка, признававшихся «священными», — латинский, греческий и древнееврейский (надпись на кресте при распятии риста по евангельскому рассказу была сделана на этих трех языках). Для образования слова азот были взяты начальные и конечные буквы алфавитов этих трех языков (а, альфа, алеф и зэт, омега, тов — АААZОТ).
Составители новой химической номенклатуры 1787 г., и прежде всего инициатор ее создания Гитон де Морво, хорошо знали о существовании с древних времен слова азот. Морво отметил в «Методической энциклопедии» (1786) алхимическое значение этого термина. После опубликования «Метода химической номенклатуры» противники кислородной теории — флогистики — выступили с резкой критикой новой номенклатуры. Особенно, как отмечает сам Лавуазье в своем учебнике химии, критиковалось принятие «древних наименований». В частности, Ламетри — издатель журнала «Observations sur la Physique» — оплота противников кислородной теории, указывал на то, что слово азот употреблялось алхимиками в другом смысле.
Несмотря на это, новое название было принято во Франции, а также и в России, заменив собою ранее принятые названия «флогистированный газ», «мофетт», «основание мофетта» и т. д.
Словообразование азот от греческого тоже вызвало справедливые замечания. Д. Н. Прянишников в своей книге «Азот в жизни растений и в земледелии СССР» (1945) совершенно правильно заметил, что словообразование от греческого «вызывает сомнения». Очевидно, эти сомнения имелись и у современников Лавуазье. Сам Лавуазье в своем учебнике химии (1789) употребляет слово азот наряду с названием «радикал нитрик» (radical nitrique).
Интересно отметить, что более поздние авторы, пытаясь, видимо, как-то оправдать неточность, допущенную членами номенклатурной комиссии, производили слово азот от греческого — дающий жизнь, животворный, создав искусственное слово «азотикос», отсутствующее в греческом языке (Диргарт, Реми и др.). Одиако этот путь образования слова азот едва ли может быть признан правильным, так как производное слово для названия азот должно было бы звучать «азотикон».
Неудачность названия азот была очевидной для многих современников Лавуазье, вполне сочувствовавших его кислородной теории. Так, Шапталь в своем учебнике химии «Элементы химии» (1790) предложил заменить слово азот словом нитроген (нитрожен) и называл газ, соответственно воззрениям своего времени (каждая молекула газа представлялась окруженной атмосферой теплорода), «газ нитрожен» (Gas nitrogene). Свое предложение Шапталь подробно мотивировал. Одним из доводов послужило указание, что название, означающее безжизненный, могло бы с большими основаниями быть дано другим простым телам (обладающим, например, сильными ядовитыми свойствами).
Название нитроген, принятое в Англии и в Америке, стало в дальнейшем основой международного названия элемента (Nitrogenium) и символа азота — N. Во Франции в начале Х1Х в. вместо символа -N употребляли символ Az. В 1800 г. один из соавторов химической номенклатуры — Фуркруа предложил еще одно название — алкалиген (алкалижен — alcaligene), исходя из того, что азот является «основанием» летучей щелочи (Alcali volatil) аммиака. Но это название не было принято химиками. Упомянем, наконец, название азота, которое употребляли химики-флогистики и, в частности Пристлей, в конце ХVIII в.- септон (Septon от французского Septique- гнилостный). Это название предложено, по-видимому, Митчелом — учеником Блэка, впоследствии работавшим в Америке. Дэви отверг это название. В Германии с конца ХVIII в. и до настоящего времени азот называют Stickstoff, что означает «удушливое вещество».
Что касается старых русских названий азота, фигурировавших в разнообразных сочинениях конца XVIII — начала ХIХ в., то они таковы: удушливый гас, нечистый гас; мофетический воздух (все это переводы французского названия Gas mofette), удушливое вещество (перевод немецкого Stickstoff), флогистированный воздух, гас огорюченный, огорюченный воздух (флогистические названия — перевод термина, предложенного Пристлеем — Рlogisticated air). Употреблялись также названия; испорченный воздух (перевод термина Шееле Verdorbene Luft), селитротвор, селитротворный гас, нитроген (перевод названия, предложенного Шапталем — Nitrogene), алкалиген, щелочетвор (термины Фуркруа, переведенные на русский язык в 1799 и 1812 гг.), септон, гнилотвор (Septon) и др. Наряду с этими многочисленными названиями употреблялись и слова азот и азотический гас, особенно с начала ХIХ в.
В.Севергин в своем «Руководстве к удобнейшему разумению химических книг иностранных» (1815) объясняет слово азот следующим образом: «Azoticum, Azotum, Azotozum — азот, удушливое вещество»; «Azote — Азот, селитротвор»; «селитротворный газ, азотовый газ». Окончательно слово азот вошло в русскую химическую номенклатуру и вытеснило все другие названия после выхода в свет «Оснований чистой химии» Г. Гесса (1831).
Производные названия соединений, содержащих азот, образованы на русском и других языках либо от слова азот (азотная кислота, азосоединения и др.), либо от международного названия нитрогениум (нитраты, нитросоединения и др.). Последний термин происходит от древних названий нитр, нитрум, нитрон, обозначавших обычно селитру, иногда — природную соду. В словаре Руланда (1612) сказано: «Нитрум, борах (baurach), селитра (Sal petrosum), нитрум, у немцев — Salpeter, Вегgsalz — то же, что и Sal реtrae».
1. Положение азота в периодической системе химических элементов
2. Строение атома азота
3. Физические свойства и нахождение в природе
4. Строение молекулы
5. Соединения азота
6. Способы получения
7. Химические свойства
7.1. Взаимодействие с простыми веществами
7.1.1. Взаимодействие с галогенами
7.1.2. Взаимодействие с серой и кремнием
7.1.3. Взаимодействие с водородом и фосфором
7.1.4. Взаимодействие с азотом
7.1.5. Взаимодействие с активными металлами
Аммиак
1. Строение молекулы и физические свойства
2. Способы получения
3. Химические свойства
3.1. Взаимодействие с серной кислотой
3.2. Взаимодействие с азотной кислотой
3.3. Взаимодействие с солями
Соли аммония
Способы получения солей аммония
Химические свойства солей аммония
Оксиды азота
1. Оксид азота (I)
2. Оксид азота (II)
3. Оксид азота (III)
4. Оксид азота (IV)
5. Оксид азота (V)
Азотная кислота
1. Строение молекулы и физические свойства
2. Способы получения
3. Химические свойства
3.1. Диссоциация азотной кислоты
2.3. Взаимодействие с основными и амфотерными оксидами и гидроксидами
2.4. Вытеснение более слабых кислот из солей
2.5. Взаимодействие с металлами
2.6. Взаимодействие с неметаллами
2.7. Окисление сложных веществ
2.8. Взаимодействие с белками
Азотистая кислота
Соли азотной кислоты — нитраты
Соли азотистой кислоты — нитриты
Азот
Положение в периодической системе химических элементов
Азот расположен в главной подгруппе V группы (или в 15 группе в современной форме ПСХЭ) и во втором периоде периодической системы химических элементов Д.И. Менделеева.
Электронное строение азота
Электронная конфигурация азота в основном состоянии:
![]()
Атом азота содержит на внешнем энергетическом уровне 3 неспаренных электрона и одну неподеленную электронную пару в основном энергетическом состоянии. Следовательно, атом азота может образовать 3 связи по обменному механизму и 1 связь по донорно-акцепторному механизму. Таким образом, максимальная валентность азота в соединениях равна IV. Также характерная валентность азота в соединениях — III.
Степени окисления атома азота – от -3 до +5. Характерные степени окисления азота -3, 0, +1, +2, +3, +4, +5.
Физические свойства и нахождение в природе
Азот в природе существует в виде простого вещества газа N2. Нет цвета, запаха и вкуса. Молекула N2 неполярная, следовательно, в воде азот практически нерастворим.
Азот – это основной компонент воздуха (79% по массе). В земной коре азот встречается в основном в виде нитратов. Входит в состав белков, аминокислот и нуклеиновых кислот в живых организмах.
Строение молекулы
Связь между атомами в молекуле азота – тройная, т.к. у каждого атома в молекуле по 3 неспаренных электрона. Одна σ-связь (сигма-связь) и две — π-связи.
Структурная формула молекулы азота:

Структурно-графическая формула молекулы азота: N≡N.
Схема перекрывания электронных облаков при образовании молекулы азота:

Соединения азота
Типичные соединения азота:
| Степень окисления | Типичные соединения |
| +5 | оксид азота (V) N2O5 азотная кислота HNO3 нитраты MeNO3 |
| +4 | оксид азота (IV) NO2 |
| +3 | оксид азота (III) азотистая кислота нитриты MeNO2 |
| +2 | оксид азота (II) NO |
| +1 | оксид азота (I) |
| -3 | аммиак NH3 нитриды металлов MeN бинарные соединения азота с неметаллами |
Способы получения азота
1. Азот в лаборатории получают при взаимодействии насыщенных растворов хлорида аммония и нитрита натрия. Образующийся в результате реакции обмена нитрит аммония легко разлагается с образованием азота и воды. В колбу наливают раствор хлорида аммония, а капельную воронку раствор нитрита натрия. При приливании нитрита натрия в колбу начинается выделение азота. Собирают выделяющийся азот в цилиндр. Горящая лучинка в атмосфере азота гаснет.
NaNO2 + NH4Cl → NH4NO2 + NaCl
NH4NO2 → N2 + 2H2O
Суммарное уравнение процесса:
NaNO2 + NH4Cl → N2 + NaCl + 2H2O
Видеоопыт взаимодействия нитрита натрия с хлоридом аммония можно посмотреть здесь.
Азот также образуется при горении аммиака:
4NH3 + 3O2 → 2N2 + 6H2O
2. Наиболее чистый азот получают разложением азидов щелочных металлов.
Например, разложением азида натрия:
2NaN3 → 2Na + 3N2
3. Еще один лабораторный способ получения азота — восстановление оксида меди (II) аммиаком при температуре ~700 °C:
3CuO + 2NH3 → 3Cu + N2 + 3H2O
В промышленности азот получают, буквально, из воздуха. При промышленном производстве очень важно, чтобы сырье было дешевым и доступным. Воздуха много и он пока бесплатный.
Используются различные способы выделения азота из воздуха — адсорбционная технология, мембранная и криогенная технологии.
Адсорбционные методы разделения воздуха на компоненты основаны на разделения газовых сред в азотных установках лежит явление связывания твёрдым веществом, называемым адсорбентом, отдельных компонентов газовой смеси.
Основным принципом работы мембранных систем является разница в скорости проникновения компонентов газа через вещество мембраны. Движущей силой разделения газов является разница парциальных давлений на различных сторонах мембраны.
В основе работы криогенных установок разделения воздуха лежит метод разделения газовых смеси, основанный на разности температур кипения компонентов воздуха и различии составов находящихся в равновесии жидких и паровых смесей.
Химические свойства азота
При нормальных условиях азот химически малоактивен.
1. Азот проявляет свойства окислителя (с элементами, которые расположены ниже и левее в Периодической системе) и свойства восстановителя (с элементами, расположенными выше и правее). Поэтому азот реагирует с металлами и неметаллами.
1.1. Молекулярный азот при обычных условиях с кислородом не реагирует. Реагирует с кислородом только при высокой температуре (2000оС), на электрической дуге (в природе – во время грозы):
N2 + O2 ⇄ 2NO – Q
Процесс эндотермический, т.е. протекает с поглощением теплоты.
1.2. При сильном нагревании (3000оС-5000оС или действие электрического разряда) образуется атомарный азот, который реагирует с серой, фосфором, мышьяком, углеродом с образованием бинарных соединений:
2С + N2 → N≡C–C≡N
Молекулярный азот, таким образом, не реагирует с серой, фосфором, мышьяком, углеродом.
1.3. Азот взаимодействует с водородом при высоком давлении и высокой температуре, в присутствии катализатора. При этом образуется аммиак:
N2 + ЗН2 ⇄ 2NH3
Этот процесс экзотермический, т.е. протекает с выделением теплоты.
1.4. Азот реагирует с активными металлами: с литием при комнатной температуре, кальцием, натрием и магнием при нагревании. При этом образуются бинарные соединения-нитриды.
Например, литий реагирует с азотом с образованием нитрида лития:
N2 + 6Li → 2Li3N
2. Со сложными веществами азот практически не реагирует из-за крайне низкой реакционной способности.
Взаимодействие возможно только в жестких условиях с активными веществами, например, сильными восстановителями.
Например, азот окисляет гидрид лития:
N2 + 3LiH → Li3N + NH3
Аммиак
Строение молекулы и физические свойства
В молекуле аммиака NH3 атом азота соединен тремя одинарными ковалентными полярными связями с атомами водорода:

Геометрическая форма молекулы аммиака — правильная треугольная пирамида. Валентный угол H-N-H составляет 107,3о:

У атома азота в аммиаке на внешнем энергетическом уровне остается одна неподеленная электронная пара. Эта электронная пара оказывает значительное влиение на свойства аммиака, а также на его структуру. Электронная структура аммиака — тетраэдр , с атомом азота в центре:

Аммиак – бесцветный газ с резким характерным запахом. Ядовит. Весит меньше воздуха. Связь N-H — сильно полярная, поэтому между молекулами аммиака в жидкой фазе возникают водородные связи. При этом аммиак очень хорошо растворим в воде, т.к. молекулы аммиака образуют водородные связи с молекулами воды.
Способы получения аммиака
В лаборатории аммиак получают при взаимодействии солей аммония с щелочами. Поск
ольку аммиак очень хорошо растворим в воде, для получения чистого аммиака используют твердые вещества.
Например, аммиак можно получить нагреванием смеси хлорида аммония и гидроксида кальция. При нагревании смеси происходит образование соли, аммиака и воды:
2NH4Cl + Са(OH)2 → CaCl2 + 2NH3 + 2Н2O
Тщательно растирают ступкой смесь соли и основания и нагревают смесь. Выделяющийся газ собирают в пробирку (аммиак — легкий газ и пробирку нужно перевернуть вверх дном). Влажная лакмусовая бумажка синеет в присутствии аммиака.
Видеоопыт получения аммиака из хлорида аммония и гидроксида кальция можно посмотреть здесь.
Еще один лабораторный способ получения аммиака – гидролиз нитридов.
Например, гидролиз нитрида кальция:
Ca3N2 + 6H2O → ЗСа(OH)2 + 2NH3
В промышленности аммиак получают с помощью процесса Габера: прямым синтезом из водорода и азота.
N2 + 3Н2 ⇄ 2NH3
Процесс проводят при температуре 500-550оС и в присутствии катализатора. Для синтеза аммиака применяют давления 15-30 МПа. В качестве катализатора используют губчатое железо с добавками оксидов алюминия, калия, кальция, кремния. Для полного использования исходных веществ применяют метод циркуляции непровзаимодействовавших реагентов: не вступившие в реакцию азот и водород вновь возвращают в реактор.
Более подробно про технологию производства аммиака можно прочитать здесь.
Химические свойства аммиака
1. В водном растворе аммиак проявляет основные свойства (за счет неподеленной электронной пары). Принимая протон (ион H+), он превращается в ион аммония. Реакция может протекать и в водном растворе, и в газовой фазе:
:NH3 + H2O ⇄ NH4+ + OH–

Таким образом, среда водного раствора аммиака – щелочная. Однако аммиак – слабое основание. При 20 градусах один объем воды поглощает до 700 объемов аммиака.
Видеоопыт растворения аммиака в воде можно посмотреть здесь.
2. Как основание, аммиак взаимодействует с кислотами в растворе и в газовой фазе с образованием солей аммония.
Например, аммиак реагирует с серной кислотой с образованием либо кислой соли – гидросульфата аммония (при избытке кислоты), либо средней соли – сульфата аммония (при избытке аммиака):
NH3 + H2SO4 → NH4HSO4
2NH3 + H2SO4 → (NH4)2SO4
Еще один пример: аммиак взаимодействует с водным раствором углекислого газа с образованием карбонатов или гидрокарбонатов аммония:
NH3 + H2O + CO2 → NH4HCO3
2NH3 + H2O + CO2 → (NH4)2CO3
Видеоопыт взаимодействия аммиака с концентрированными кислотами – азотной, серной и и соляной можно посмотреть здесь.
В газовой фазе аммиак реагирует с летучим хлороводородом. При этом образуется густой белый дым – это выделяется хлорид аммония.
NH3 + HCl → NH4Cl
Видеоопыт взаимодействия аммиака с хлороводородом в газовой фазе (дым без огня) можно посмотреть здесь.
3. В качестве основания, водный раствор аммиака реагирует с растворами солей тяжелых металлов, образуя нерастворимые гидроксиды.
Например, водный раствор аммиака реагирует с сульфатом железа (II) с образованием сульфата аммония и гидроксида железа (II):
FeSO4 + 2NH3 + 2H2O → Fe(OH)2 + (NH4)2SO4
4. Соли и гидроксиды меди, никеля, серебра растворяются в избытке аммиака, образуя комплексные соединения – амминокомплексы.
Например, хлорид меди (II) реагирует с избытком аммиака с образованием хлорида тетрамминомеди (II):
4NH3 + CuCl2 → [Cu(NH3)4]Cl2
Гидроксид меди (II) растворяется в избытке аммиака:
4NH3 + Cu(OH)2 → [Cu(NH3)4](OH)2
5. Аммиак горит на воздухе, образуя азот и воду:
4NH3 + 3O2 → 2N2 + 6H2O
Если реакцию проводить в присутствии катализатора (Pt), то азот окисляется до NO:
4NH3 + 5O2 → 4NO + 6H2O
6. За счет атомов водорода в степени окисления +1 аммиак может выступать в роли окислителя, например в реакциях с щелочными, щелочноземельными металлами, магнием и алюминием. С металлами реагирует только жидкий аммиак.
Например, жидкий аммиак реагирует с натрием с образованием амида натрия:
2NH3 + 2Na → 2NaNH2 + H2
Также возможно образование Na2NH, Na3N.
При взаимодействии аммиака с алюминием образуется нитрид алюминия:
2NH3 + 2Al → 2AlN + 3H2
7. За счет азота в степени окисления -3 аммиак проявляет восстановительные свойства. Может взаимодействовать с сильными окислителями — хлором, бромом, пероксидом водорода, пероксидами и оксидами некоторых металлов. При этом азот окисляется, как правило, до простого вещества.
Например, аммиак окисляется хлором до молекулярного азота:
2NH3 + 3Cl2 → N2 + 6HCl
Пероксид водорода также окисляет аммиак до азота:
2NH3 + 3H2O2 → N2 + 6H2O
Оксиды металлов, которые в электрохимическом ряду напряжений металлов расположены справа — сильные окислители. Поэтому они также окисляют аммиак до азота.
Например, оксид меди (II) окисляет аммиак:
2NH3 + 3CuO → 3Cu + N2 + 3H2O
Соли аммония
Соли аммония – это соли, состоящие из катиона аммония и аниона кислотного остатка.
Способы получения солей аммония
1. Соли аммония можно получить взаимодействием аммиака с кислотами. Реакции подробно описаны выше.
2. Соли аммония также получают в обменных реакциях между солями аммония и другими солями.
Например, хлорид аммония реагирует с нитратом серебра:
NH4Cl + AgNO3 → AgCl + NH4NO3
3. Средние соли аммония можно получить из кислых солей аммония. При добавлении аммиака кислая соль переходит в среднюю.
Например, гидрокарбонат аммония реагирует с аммиаком с образованием карбоната аммония:
NH4НCO3 + NH3 → (NH4)2CO3
Химические свойства солей аммония
1. Все соли аммония – сильные электролиты, почти полностью диссоциируют на ионы в водных растворах:
NH4Cl ⇄ NH4+ + Cl–
2. Соли аммония проявляют свойства обычных растворимых солей –вступают в реакции обмена с щелочами, кислотами и растворимыми солями, если в продуктах образуется газ, осадок или образуется слабый электролит.
Например, карбонат аммония реагирует с соляной кислотой. При этом выделяется углекислый газ:
(NH4)2CO3 + 2НCl → 2NH4Cl + Н2O + CO2
Соли аммония реагируют с щелочами с образованием аммиака.
Например, хлорид аммония реагирует с гидроксидом калия:
NH4Cl + KOH → KCl + NH3 + H2O
Взаимодействие с щелочами — качественная реакция на ионы аммония. Выделяющийся аммиак можно обнаружить по характерному резкому запаху и посинению лакмусовой бумажки.
3. Соли аммония подвергаются гидролизу по катиону, т.к. гидроксид аммония — слабое основание:
NH4Cl + Н2O ↔ NH3 ∙ H2O + HCl
NH4+ + HOH ↔ NH3 ∙ H2O + H+
4. При нагревании соли аммония разлагаются. При этом если соль не содержит анион-окислителя, то разложение проходит без изменения степени окисления атома азота. Так разлагаются хлорид, карбонат, сульфат, сульфид и фосфат аммония:
NH4Cl → NH3 + HCl
NH4HCO3 → NH3 + CO2 + H2O
(NH4)2SO4 → NH4HSO4 + NH3
NH4HS → NH3 + H2S
Если соль содержит анион-окислитель, то разложение сопровождается изменением степени окисления атома азота иона аммония. Так протекает разложение нитрата, нитрита и дихромата аммония:
NH4NO2 → N2 + 2H2O
190 – 245° C:
NH4NO3 → N2O + 2H2O
При температуре 250 – 300°C:
2NH4NO3 → 2NO + 4H2O
При температуре выше 300°C:
2NH4NO3 → 2N2 + O2 + 4H2O
Разложение бихромата аммония («вулканчик»). Оранжевые кристаллы дихромата аммония под действием горящей лучинки бурно реагируют. Дихромат аммония – особенная соль, в ее составе – окислитель и восстановитель. Поэтому «внутри» этой соли может пройти окислительно-восстановительная реакция (внутримолекулярная ОВР):
(NH4)2Cr2O7 → Cr2O3 + N2 + 4H2O
Окислитель – хром (VI) превращается в хром (III), образуется зеленый оксид хрома. Восстановитель – азот, входящий в состав иона аммония, превращается в газообразный азот. Итак, дихромат аммония превращается в зеленый оксид хрома, газообразный азот и воду. Реакция начинается от горящей лучинки, но не прекращается, если лучинку убрать, а становится еще интенсивней, так как в процессе реакции выделяется теплота, и, начавшись от лучинки, процесс лавинообразно развивается. Оксид хрома (III) – очень твердое, тугоплавкое вещество зеленого цвета, его используют как абразив. Температура плавления – почти 2300 градусов. Оксид хрома – очень устойчивое вещество, не растворяется даже в кислотах. Благодаря устойчивости и интенсивной окраске окись хрома используется при изготовлении масляных красок.
Видеоопыт разложения дихромата аммония можно посмотреть здесь.
Оксиды азота
| Оксиды азота | Цвет | Фаза | Характер оксида |
| N2O Оксид азота (I), закись азота, «веселящий газ» | бесцветный | газ | несолеобразующий |
| NO Оксид азота (II) | бесцветный | газ | несолеобразующий |
| N2O3 Оксид азота (III), азотистый ангидрид | синий | жидкость | кислотный |
| NO2 Оксид азота (IV), диоксид азота, «лисий хвост» | бурый | газ | кислотный (соответствуют две кислоты) |
| N2O5 Оксид азота (V), азотный ангидрид | бесцветный | твердый | кислотный |
Оксид азота (I)
Оксид азота (I) – это несолеобразующий оксид. Малые концентрации закиси азота вызывают лёгкое опьянение (отсюда название — «веселящий газ»). При вдыхании чистого газа быстро развиваются состояние опьянения и сонливость. Закись азота обладает слабой наркотической активностью, в связи с чем в медицине её применяют в больших концентрациях. В смеси с кислородом при правильном дозировании (до 80 % закиси азота) вызывает хирургический наркоз.
Строение молекулы оксида азота (I) нельзя описать методом валентных связей. Так как оксид азота (I) состоит из двух, так называемых резонансных структур, которые переходят одна в другую:
![]()
Общую формулу в таком случае можно задать, обозначая изменяющиеся связи в резонансных структурах пунктиром:
![]()
Получить оксид азота (I) в лаборатории можно разложением нитрата аммония:
NH4NO3 → N2O + 2H2O
Химические свойства оксида азота (I):
1. При нормальных условиях оксид азота (I) инертен. При нагревании проявляет свойства окислителя. Оксид азота (I) при нагревании окисляет водород, аммиак, металлы, сернистый газ и др. При этом азот восстанавливается в простое вещество.
N2O + H2 → N2 + H2O
N2O + Mg → N2 + MgO
N2O + 2Cu → N2 + Cu2O
3N2O + 2NH3 → 4N2 + 3H2O
N2O + H2O + SO2 → N2 + H2SO4
Еще пример: оксид азота (I) окисляет углерод и фосфор при нагревании:
N2O + C → N2 + CO
5N2O + 2Р → 5N2 + Р2O5
2. При взаимодействии с сильными окислителями N2O может проявлять свойства восстановителя.
Например, N2O окисляется раствором перманганата в серной кислоте:
5N2O + 3H2SO4 + 2KMnO4 → 10NO + 2MnSO4 + K2SO4 + 3H2O
Оксид азота (II)
Оксид азота (II) – это несолеобразующий оксид. В нормальных условиях это бесцветный ядовитый газ, плохо растворимый в воде. На воздухе коричневеет из-за окисления до диоксида азота. Сжижается с трудом; в жидком и твёрдом виде имеет голубой цвет.
Способы получения.
1. В лаборатории оксид азота (II) получают действием разбавленной азотной кислоты (30%) на неактивные металлы.
Например, при действии 30 %-ной азотной кислоты на медь образуется NO:
3Cu + 8HNO3(разб.) → 3Cu(NO3)2 + 2NO + 4H2O
Также NO можно получить при окислении хлорида железа (II) или иодоводорода азотной кислотой:
3FeCl2 + NaNO3 + 4HCl → 3FeCl3 + NaCl + NO + 2H2O
2HNO3 + 6HI → 2NO + I2 + 4H2O
2. В природе оксид азота (II) образуется из азота и кислорода под действием электрического разряда, например, во время грозы:
N2 + O2 → 2NO
3. В промышленности оксид азота (II) получают каталитическим окислением аммиака:
4NH3 + 5O2 → 4NO + 6H2O
Химические свойства.
1. Оксид азота (II) легко окисляется под действием окислителей.
Например, горит в атмосфере кислорода:
2NO + O2 → 2NO2
Оксид азота (II) легко окисляется под действием хлора или озона:
2NO + Cl2 → 2NOCl
NO + O3 → NO2 + O2
2. В присутствии более сильных восстановителей проявляет свойства окислителя. В атмосфере оксида азота (II) могут гореть водород, углерод и т.п.
Например, оксид азота (II) окисляет водород и сернистый газ:
2NO + 2H2 → N2 + 2H2O
2NO + 2SO2 → 2SO3 + N2
Оксид азота (III)
Оксид азота (III), азотистый ангидрид – кислотный оксид. За счет азота со степенью окисления +3 проявляет восстановительные и окислительные свойства. Устойчив только при низких температурах, при более высоких температурах разлагается.
Способы получения: можно получить при низкой температуре из оксидов азота:
NO2 + NO ↔ N2O3
Химические свойства:
1. Оксид азота (III) взаимодействует с водой с образованием азотистой кислоты:
N2O3 + H2O ↔ 2HNO2
2. Оксид азота (III) взаимодействует с основаниями и основными оксидами:
Например, оксид азота (III) реагирует с гидроксидом и оксидом натрия с образованием нитрита натрия и воды:
N2O3 + 2NaOH → 2NaNO2 + H2O
N2O3 + Na2O → 2NaNO2
Оксид азота (IV)
Оксид азота (IV) — бурый газ. Очень ядовит! Для NO2 характерна высокая химическая активность.
Способы получения.
1. Оксид азота (IV) образуется при окислении оксида азота (I) и оксида азота (II) кислородом или озоном:
2NO + O2 → 2NO2
2. Оксид азота (IV) образуется при действии концентрированной азотной кислоты на неактивные металлы.
Например, при действии концентрированной азотной кислоты на медь:
4HNO3(конц.) + Cu → Cu(NO3)2 + 2NO2 + 2H2O
3. Оксид азота (IV) образуется также при разложении нитратов металлов, которые в ряду электрохимической активности расположены правее магния (включая магний) и при разложении нитрата лития.
Например, при разложении нитрата серебра:
2AgNO3 → 2Ag + 2NO2 + O2
Химические свойства.
1. Оксид азота (IV) реагирует с водой с образованием двух кислот — азотной и азотистой:
2NO2 + H2O → HNO3 + HNO2
Если растворение NO2 в воде проводить в избытке кислорода, то образуется только азотная кислота:
4NO2 + 2H2O + O2 → 4HNO3
Поскольку азотистая кислота неустойчива, то при растворении NO2 в теплой воде образуются HNO3 и NO:
3NO2 + H2O → 2HNO3 + NO
2. При растворении оксида азота (IV) в щелочах образуются нитраты и нитриты:
2NO2 + 2NaOH → NaNO3 + NaNO2 + H2O
4NO2 + 2Ca(OH)2 → Ca(NO2)2 + Ca(NO3)2 + 2H2O
В присутствии кислорода образуются только нитраты:
4NO2 + 4NaOH + O2 → 4NaNO3 + 2H2O
3. Оксид азота (IV) – сильный окислитель. В атмосфере оксида азота (IV) горят фосфор, уголь, сера, оксид серы (IV) окисляется до оксида серы (VI):
2NO2 + 2S → N2 + 2SO2
2NO2 + 2C → N2 + 2CO2
10NO2 + 8P → 5N2 + 4P2O5
NO2 + SO2 → SO3 + NO
4. Оксид азота (IV) димеризуется:
2NO2 ⇄ N2O4
Оксид азота (V)
N2O5 – оксид азота (V), ангидрид азотной кислоты – кислотный оксид.
Получение оксида азота (V).
1. Получить оксид азота (V) можно окислением диоксида азота:
2NO2 + O3 → N2O5 + O2
2. Еще один способ получения оксида азота (V) – обезвоживание азотной кислоты сильным водоотнимающим веществом, оксидом фосфора (V):
2HNO3 + P2O5 → 2HPO3 + N2O5
Химические свойства оксида азота (V).
1. При растворении в воде оксид азота (V) образует азотную кислоту:
N2O5 + H2O → 2HNO3
2. Оксид азота (V), как типичный кислотный оксид, взаимодействует с основаниями и основными оксидами с образованием солей-нитратов.
Например, оксид азота (V) реагирует с гидроксидом натрия:
N2O5 + 2NaOH → 2NaNO3 + H2O
Еще пример: оксид азота (V) реагирует с оксидом кальция:
N2O5 + CaO → Ca(NO3)2
3. За счет азота со степенью окисления +5 оксид азота (V) – сильный окислитель.
Например, он окисляет серу:
2N2O5 + S → SO2 + 4NO2
4. Оксид азота (V) легко разлагается при нагревании (со взрывом):
2N2O5 → 4NO2 + O2
Азотная кислота
Строение молекулы и физические свойства
Азотная кислота HNO3 – это сильная одноосновная кислота-гидроксид. При обычных условиях бесцветная, дымящая на воздухе жидкость, температура плавления −41,59 °C, кипения +82,6 °C ( при нормальном атмосферном давлении). Азотная кислота смешивается с водой во всех соотношениях. На свету частично разлагается.
Валентность азота в азотной кислоте равна IV, так как валентность V у азота отсутствует. При этом степень окисления атома азота равна +5. Так происходит потому, что атом азота образует 3 обменные связи и одну донорно-акцепторную, является донором электронной пары.
Поэтому строение молекулы азотной кислоты можно описать резонансными структурами:


Обозначим дополнительные связи между азотом и кислородом пунктиром. Этот пунктир по сути обозначает делокализованные электроны. Получается формула:

Способы получения
В лаборатории азотную кислоту можно получить разными способами:
1. Азотная кислота образуется при действии концентрированной серной кислоты на твердые нитраты металлов. При этом менее летучая серная кислота вытесняет более летучую азотную.
Например, концентрированная серная кислота вытесняет азотную из кристаллического нитрата калия:
KNO3 + H2SO4(конц) → KHSO4 + HNO3
2. В промышленности азотную кислоту получают из аммиака. Процесс осуществляется стадийно.
1 стадия. Каталитическое окисление аммиака.
4NH3 + 5O2 → 4NO + 6H2O
2 стадия. Окисление оксида азота (II) до оксида азота (IV) кислородом воздуха.
2NO + O2 → 2NO2
3 стадия. Поглощение оксида азота (IV) водой в присутствии избытка кислорода.
4NO2 + 2H2O + O2 → 4HNO3
Химические свойства
Азотная кислота – это сильная кислота. За счет азота со степенью окисления +5 азотная кислота проявляет сильные окислительные свойства.
1. Азотная кислота практически полностью диссоциирует в водном растворе.
HNO3 → H+ + NO3–
2. Азотная кислота реагирует с основными оксидами, основаниями, амфотерными оксидами и амфотерными гидроксидами.
Например, азотная кислота взаимодействует с оксидом меди (II):
CuO + 2HNO3 → Cu(NO3)2 + H2O
Еще пример: азотная кислота реагирует с гидроксидом натрия:
HNO3 + NaOH → NaNO3 + H2O
3. Азотная кислота вытесняет более слабые кислоты из их солей (карбонатов, сульфидов, сульфитов).
Например, азотная кислота взаимодействует с карбонатом натрия:
2HNO3 + Na2CO3 → 2NaNO3 + H2O + CO2
4. Азотная кислота частично разлагается при кипении или под действием света:
4HNO3 → 4NO2 + O2 + 2H2O
5. Азотная кислота активно взаимодействует с металлами. При этом никогда не выделяется водород! При взаимодействии азотной кислоты с металлами окислителем всегда выступает азот +5. Азот в степени окисления +5 может восстанавливаться до степеней окисления -3, 0, +1, +2 или +4 в зависимости от концентрации кислоты и активности металла.
металл + HNO3 → нитрат металла + вода + газ (или соль аммония)
С алюминием, хромом и железом на холоду концентрированная HNO3 не реагирует – кислота «пассивирует» металлы, т.к. на их поверхности образуется пленка оксидов, непроницаемая для концентрированной азотной кислоты. При нагревании реакция идет. При этом азот восстанавливается до степени окисления +4:
Fe + 6HNO3(конц.) → Fe(NO3)3 + 3NO2 + 3H2O
Al + 6HNO3(конц.) → Al(NO3)3 + 3NO2 + 3H2O
Золото и платина не реагируют с азотной кислотой, но растворяются в «царской водке» – смеси концентрированных азотной и соляной кислот в соотношении 1 : 3 (по объему):
HNO3 + 3HCl + Au → AuCl3 + NO + 2H2O
Концентрированная азотная кислота взаимодействует с неактивными металлами и металлами средней активности (в ряду электрохимической активности после алюминия). При этом образуется оксид азота (IV), азот восстанавливается минимально:
4HNO3(конц.) + Cu → Cu(NO3)2 + 2NO2 + 2H2O
С активными металлами (щелочными и щелочноземельными) концентрированная азотная кислота реагирует с образованием оксида азота (I):
10HNO3 + 4Ca → 4Ca(NO3)2 + 2N2O + 5H2O
Разбавленная азотная кислота взаимодействует с неактивными металлами и металлами средней активности (в ряду электрохимической активности после алюминия). При этом образуется оксид азота (II).
8HNO3 (разб.) + 3Cu → 3Cu(NO3)2 + 2NO + 4H2O
С активными металлами (щелочными и щелочноземельными), а также оловом и железом разбавленная азотная кислота реагирует с образованием молекулярного азота:
12HNO3(разб) + 10Na → 10NaNO3 + N2 + 6H2O
При взаимодействии кальция и магния с азотной кислотой любой концентрации (кроме очень разбавленной) образуется оксид азота (I):
10HNO3 + 4Ca → 4Ca(NO3)2 + 2N2O + 5H2O
Очень разбавленная азотная кислота реагирует с металлами с образованием нитрата аммония:
10HNO3 + 4Zn → 4Zn(NO3)2 + NH4NO3 + 3H2O
Таблица. Взаимодействие азотной кислоты с металлами.
| Азотная кислота | ||||
| Концентрированная | Разбавленная | |||
| с Fe, Al, Cr | с неактивными металлами и металлами средней активности (после Al) | с щелочными и щелочноземельными металлами | с неактивными металлами и металлами средней активности (после Al) | с металлами до Al в ряду активности, Sn, Fe |
| пассивация при низкой Т | образуется NO2 | образуется N2O | образуется NO | образуется N2 |
6. Азотная кислота окисляет и неметаллы (кроме кислорода, водорода, хлора, фтора и некоторых других). При взаимодействии с неметаллами HNO3 обычно восстанавливается до NO или NO2, неметаллы окисляются до соответствующих кислот, либо оксидов (если кислота неустойчива).
Например, азотная кислота окисляет серу, фосфор, углерод, йод:
6HNO3 + S → H2SO4 + 6NO2 + 2H2O
Безводная азотная кислота – сильный окислитель. Поэтому она легко взаимодействует с красным и белым фосфором. Реакция с белым фосфором протекает очень бурно. Иногда она сопровождается взрывом.
5HNO3 + P → H3PO4 + 5NO2 + H2O
5HNO3 + 3P + 2H2O → 3H3PO4 + 5NO
Видеоопыт взаимодействия фосфора с безводной азотной кислотой можно посмотреть здесь.
4HNO3 + C → CO2 + 4NO2 + 2H2O
Видеоопыт взаимодействия угля с безводной азотной кислотой можно посмотреть здесь.
10HNO3 + I2 → 2HIO3 + 10NO2 + 4H2O
7. Концентрированная азотная кислота окисляет сложные вещества (в которых есть элементы в отрицательной, либо промежуточной степени окисления): сульфиды металлов, сероводород, фосфиды, йодиды, соединения железа (II) и др. При этом азот восстанавливается до NO2, неметаллы окисляются до соответствующих кислот (или оксидов), а металлы окисляются до устойчивых степеней окисления.
Например, азотная кислота окисляет оксид серы (IV):
2HNO3 + SO2 → H2SO4 + 2NO2
Еще пример: азотная кислота окисляет йодоводород:
6HNO3 + HI → HIO3 + 6NO2 + 3H2O
Азотная кислота окисляет углерод до углекислого газа, т.к. угольная кислота неустойчива.
3С + 4HNO3 → 3СО2 + 4NO + 2H2O
Сера в степени окисления -2 окисляется без нагревания до простого вещества, при нагревании до серной кислоты.
Например, сероводород окисляется азотной кислотой без нагревания до молекулярной серы:
2HNO3 + H2S → S + 2NO2 + 2H2O
При нагревании до серной кислоты:
2HNO3 + H2S → H2SO4 + 2NO2 + 2H2O
8HNO3 + CuS → CuSO4 + 8NO2 + 4H2O
Соединения железа (II) азотная кислота окисляет до соединений железа (III):
4HNO3 + FeS → Fe(NO3)3 + NO + S + 2H2O
8. Азотная кислота окрашивает белки в оранжево-желтый цвет («ксантопротеиновая реакция»).
Ксантопротеиновую реакцию проводят для обнаружения белков, содержащих в своем составе ароматические аминокислоты. К раствору белка прибавляем концентрированную азотную кислоту. Белок свертывается. При нагревании белок желтеет. При добавлении избытка аммиака окраска переходит в оранжевую.

Видеоопыт обнаружения белков с помощью азотной кислоты можно посмотреть здесь.
Азотистая кислота
Азотистая кислота HNO2 — слабая, одноосновная, химически неустойчивая кислота.
Получение азотистой кислоты.
Азотистую кислоту легко получить вытеснением из нитритов более сильной кислотой.
Например, соляная кислота вытесняет азотистую кислоту из нитрита серебра:
AgNO2 + HCl → HNO2 + AgCl
Химические свойства.
1. Азотистая кислота HNO2 существует только в разбавленных растворах, при нагревании она разлагается:
3HNO2 → HNO3 + 2NO + H2O
без нагревания азотистая кислота также разлагается:
2HNO2 → NO2 + NO + H2O
2. Азотистая кислота взаимодействует с сильными основаниями.
Например, с гидроксидом натрия:
HNO2 + NaOH → NaNO2 + H2O
3. За счет азота в степени окисления +3 азотистая кислота проявляет слабые окислительные свойства. Окислительные свойства HNO2 проявляет только при взаимодействии с сильными восстановителями.
Например, HNO2 окисляет иодоводород:
2HNO2 + 2HI → 2NO + I2 + 2H2O
Азотистая кислота также окисляет иодиды в кислой среде:
2НNO2 + 2KI + 2H2SO4 → K2SO4 + I2 + 2NO + 2H2O
Азотистая кислота окисляет соединения железа (II):
2HNO2 + 3H2SO4 + 6FeSO4 → 3Fe2(SO4)3 + N2 + 4H2O
4. За счет азота в степени окисления +3 азотистая кислота проявляет сильные восстановительные свойства. Под действием окислителей азотистая кислота переходит в азотную.
Например, хлор окисляет азотистую кислоту до азотной кислоты:
HNO2 + Cl2 + H2O → HNO3 + 2HCl
Кислород и пероксид водорода также окисляют азотистую кислоту:
2HNO2 + O2 → 2HNO3
HNO2 + H2O2 → HNO3 + H2O
Соединения марганца (VII) окисляют HNO2:
5HNO2 + 2HMnO4 → 2Mn(NO3)2 + HNO3 + 3H2O
Соли азотной кислоты — нитраты
Нитраты металлов — это твердые кристаллические вещества. Большинство очень хорошо растворимы в воде.
1. Нитраты термически неустойчивы, причем все они разлагаются на кислород и соединение, характер которого зависит от положения металла (входящего в состав соли) в ряду напряжений металлов:
- Нитраты щелочных и щелочноземельных металлов (до Mg в электрохимическом ряду) разлагаются до нитрита и кислорода.
Например, разложение нитрата натрия:
2KNO3 → 2KNO2 + O2
Исключение – литий.
Видеоопыт разложения нитрата калия можно посмотреть здесь.
- Нитраты тяжелых металлов (от Mg до Cu, включая магний и медь) и литий разлагаются до оксида металла, оксида азота (IV) и кислорода:
Например, разложение нитрата меди (II):
2Cu(NO3)2 → 2CuO + 4NO2 + O2
- Нитраты малоактивных металлов (правее Cu) – разлагаются до металла, оксида азота (IV) и кислорода.
Например, нитрат серебра:
2AgNO3 → 2Ag + 2NO2 + O2

Исключения:
Нитрит железа (II) разлагается до оксида железа (III):
4Fe(NO3)2 → 2Fe2O3 + 8NO2 + O2
Нитрат марганца (II) разлагается до оксида марганца (IV):
Mn(NO3)2 → MnO2 + 2NO2
2. Водные растворы не обладают окислительно-восстановительными свойствами, расплавы – сильные окислители.
Например, смесь 75% KNO3, 15% C и 10% S называют «черным порохом»:
2KNO3 + 3C + S → N2 + 3CO2 + K2S
Соли азотистой кислоты — нитриты
Соли азотистой кислоты устойчивее самой кислоты, и все они ядовиты. Поскольку степень окисления азота в нитритах равна +3, то они проявляют как окислительные свойства, так и восстановительные.
Кислород, галогены и пероксид водорода окисляют нитриты до нитратов:
2KNO2 + O2 → 2KNO3
KNO2 + H2O2 → KNO3 + H2O
KNO2 + H2O + Br2 → KNO3 + 2HBr
Лабораторные окислители — перманганаты, дихроматы — также окисляют нитриты до нитратов:
5KNO2 + 3H2SO4 + 2KMnO4 → 5KNO3 + 2MnSO4 + K2SO4 + 3H2O
3KNO2 + 4H2SO4 + K2Cr2O7 → 3KNO3 + Cr2(SO4)3 + K2SO4 + 4H2O
В кислой среде нитриты выступают в качестве окислителей.
При окислении йодидов или соединений железа (II) нитриты восстанавливаются до оксида азота (II):
2KNO2 + 2H2SO4 + 2KI → 2NO + I2 + 2K2SO4 + 2H2O
2KNO2 + 2FeSO4 + 2H2SO4 → Fe2(SO4)3 + 2NO + K2SO4 + 2H2O
При взаимодействии с очень сильными восстановителями (алюминий или цинк в щелочной среде) нитриты восстанавливаются максимально – до аммиака:
NaNO2 + 2Al + NaOH + 6H2O → 2Na[Al(OH)4] + NH3
Смесь нитратов и нитритов также проявляет окислительные свойства. Например, смесь нитрата и нитрита калия окисляет оксид хрома (III) до хромата калия:
3KNO2 + Cr2O3 + KNO3 → 2K2CrO4 + 4NO
Азот
Азо́т — элемент главной подгруппы пятой группы второго периода периодической системы химических элементов, с атомным номером 7. Обозначается символом N (лат. Nitrogenium). Простое вещество азот (CAS-номер: 7727-37-9) — достаточно инертный при нормальных условиях двухатомный газ без цвета, вкуса и запаха (формула N2), из которого на три четверти состоит земная атмосфера.
История открытия
В 1772 году Генри Кавендиш провёл следующий опыт: он многократно пропускал воздух над раскалённым углём, затем обрабатывал его щёлочью, в результате получался остаток, который Кавендиш назвал удушливым (или мефитическим) воздухом. С позиций современной химии ясно, что в реакции с раскалённым углём кислород воздуха связывался в углекислый газ, который затем поглощался щёлочью. При этом остаток газа представлял собой по большей части азот. Таким образом, Кавендиш выделил азот, но не сумел понять, что это новое простое вещество (химический элемент). В том же году Кавендиш сообщил об этом опыте Джозефу Пристли.
Пристли в это время проводил серию экспериментов, в которых также связывал кислород воздуха и удалял полученный углекислый газ, то есть также получал азот, однако, будучи сторонником господствующей в те времена теории флогистона, совершенно неверно истолковал полученные результаты (по его мнению, процесс был противоположным — не кислород удалялся из газовой смеси, а наоборот, в результате обжига воздух насыщался флогистоном; оставшийся воздух (азот) он и назвал насыщенным флогистоном, то есть флогистированным). Очевидно, что и Пристли, хотя и смог выделить азот, не сумел понять сути своего открытия, поэтому и не считается первооткрывателем азота.
Одновременно схожие эксперименты с тем же результатом проводил и Карл Шееле.
В 1772 году азот (под названием «испорченного воздуха») как простое вещество описал Даниэль Резерфорд, он опубликовал магистерскую диссертацию, где указал основные свойства азота (не реагирует со щелочами, не поддерживает горения, непригоден для дыхания). Именно Даниэль Резерфорд и считается первооткрывателем азота. Однако и Резерфорд был сторонником флогистонной теории, поэтому также не смог понять, что же он выделил. Таким образом, чётко определить первооткрывателя азота невозможно.
В дальнейшем азот был изучен Генри Кавендишем (интересен тот факт, что он сумел связать азот с кислородом при помощи разрядов электрического тока, а после поглощения оксидов азота в остатке получил небольшое количество газа, абсолютно инертного, хотя, как и в случае с азотом, не смог понять, что выделил новый химический элемент — инертный газ аргон).
Происхождение названия
Азо́т (от др.-греч. ἄζωτος — безжизненный, лат. nitrogenium), вместо предыдущих названий («флогистированный», «мефитический» и «испорченный» воздух) предложил в 1787 году Антуан Лавуазье, который в то время в составе группы других французских учёных разрабатывал принципы химической номенклатуры. Как показано выше, в то время уже было известно, что азот не поддерживает ни горения, ни дыхания. Это свойство и сочли наиболее важным. Хотя впоследствии выяснилось, что азот, наоборот, крайне необходим для всех живых существ, название сохранилось во французском и русском языках.
Существует и иная версия. Слово «азот» придумано не Лавуазье и не его коллегами по номенклатурной комиссии; оно вошло в алхимическую литературу уже в раннем средневековье и употреблялось для обозначения «первичной материи металлов», которую считали «альфой и омегой» всего сущего. Это выражение заимствовано из Апокалипсиса: «Я есмь Альфа и Омега, начало и конец» (Откр.1:8-10). Слово составлено из начальных и конечных букв алфавитов трёх языков — латинского, греческого и древнееврейского, — считавшихся «священными», поскольку, согласно Евангелиям, надпись на кресте при распятии Христа была сделана на этих языках (а, альфа, алеф и зет, омега, тав — AAAZOTH). Составители новой химической номенклатуры хорошо знали о существовании этого слова; инициатор её создания Гитон де Морво отмечал в своей «Методической энциклопедии» (1786) алхимическое значение термина.
Возможно, слово «азот» произошло от одного из двух арабских слов — либо от слова «аз-зат» («сущность» или «внутреннюю реальность»), либо от слова «зибак» («ртуть»)..
На латыни азот называется «nitrogenium», то есть «рождающий селитру»; английское название производится от латинского. В немецком языке используется название Stickstoff, что означает «удушающее вещество».
Получение
В лабораториях его можно получать по реакции разложения нитрита аммония:
NH4NO2 → N2↑ + 2H2O
Реакция экзотермическая, идёт с выделением 80 ккал (335 кДж), поэтому требуется охлаждение сосуда при её протекании (хотя для начала реакции требуется нагревание нитрита аммония).
Практически эту реакцию выполняют, добавляя по каплям насыщенный раствор нитрита натрия в нагретый насыщенный раствор сульфата аммония, при этом образующийся в результате обменной реакции нитрит аммония мгновенно разлагается.
Выделяющийся при этом газ загрязнён аммиаком, оксидом азота (I) и кислородом, от которых его очищают, последовательно пропуская через растворы серной кислоты, сульфата железа (II) и над раскалённой медью. Затем азот осушают.
Ещё один лабораторный способ получения азота — нагревание смеси дихромата калия и сульфата аммония (в соотношении 2:1 по массе). Реакция идёт по уравнениям:
K2Cr2O7 + (NH4)2SO4 = (NH4)2Cr2O4 + K2SO4
(NH4)2Cr2O7 →(t) Cr2O3 + N2↑ + 4H2O
Самый чистый азот можно получить разложением азидов металлов:
2NaN3 →(t) 2Na + 3N2↑
Так называемый «воздушный», или «атмосферный» азот, то есть смесь азота с благородными газами, получают путём реакции воздуха с раскалённым коксом:
O2+ 4N2 + 2C → 2CO + 4N2
При этом получается так называемый «генераторный», или «воздушный», газ — сырьё для химических синтезов и топливо. При необходимости из него можно выделить азот, поглотив монооксид углерода.
Молекулярный азот в промышленности получают фракционной перегонкой жидкого воздуха. Этим методом можно получить и «атмосферный азот». Также широко применяются азотные установки и станции, в которых используется метод адсорбционного и мембранного газоразделения.
Один из лабораторных способов — пропускание аммиака над оксидом меди (II) при температуре ~700 °C:
2NH3 + 3CuO → N2↑ + 3H2O + 3Cu
Аммиак берут из его насыщенного раствора при нагревании. Количество CuO в 2 раза больше расчётного. Непосредственно перед применением азот очищают от примеси кислорода и аммиака пропусканием над медью и её оксидом (II) (тоже ~700 °C), затем сушат концентрированной серной кислотой и сухой щёлочью. Процесс происходит довольно медленно, но он того стоит: газ получается весьма чистый.
Физические свойства
При нормальных условиях азот это бесцветный газ, не имеет запаха, мало растворим в воде (2,3 мл/100г при 0 °C, 0,8 мл/100 г при 80 °C), плотность 1,2506 кг/м³ (при н.у.).
В жидком состоянии (темп. кипения −195,8 °C) — бесцветная, подвижная, как вода, жидкость. Плотность жидкого азота 808 кг/м³. При контакте с воздухом поглощает из него кислород.
При −209,86 °C азот переходит в твердое состояние в виде снегоподобной массы или больших белоснежных кристаллов. При контакте с воздухом поглощает из него кислород, при этом плавится, образуя раствор кислорода в азоте.
Источник: Википедия
Другие заметки по химии