This article is about the metallic element. For other uses, see Iron (disambiguation).
 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Iron | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allotropes | see Allotropes of iron | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Appearance | lustrous metallic with a grayish tinge | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Standard atomic weight Ar°(Fe) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Iron in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 26 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Group | group 8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Period | period 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Block | d-block | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electron configuration | [Ar] 3d6 4s2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 8, 14, 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Physical properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phase at STP | solid | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Melting point | 1811 K (1538 °C, 2800 °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Boiling point | 3134 K (2862 °C, 5182 °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density (near r.t.) | 7.874 g/cm3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| when liquid (at m.p.) | 6.98 g/cm3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of fusion | 13.81 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of vaporization | 340 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar heat capacity | 25.10 J/(mol·K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Vapor pressure
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxidation states | −4, −2, −1, 0, +1,[2] +2, +3, +4, +5,[3] +6, +7[4] (an amphoteric oxide) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 1.83 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ionization energies |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic radius | empirical: 126 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Covalent radius | Low spin: 132±3 pm High spin: 152±6 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Van der Waals radius | 194 [1] pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Spectral lines of iron | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Natural occurrence | primordial | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure | body-centered cubic (bcc)
a=286.65 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure | face-centered cubic (fcc)
between 1185–1667 K; a=364.680 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Speed of sound thin rod | 5120 m/s (at r.t.) (electrolytic) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal expansion | 11.8 µm/(m⋅K) (at 25 °C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal conductivity | 80.4 W/(m⋅K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrical resistivity | 96.1 nΩ⋅m (at 20 °C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Curie point | 1043 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Magnetic ordering | ferromagnetic | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Young’s modulus | 211 GPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Shear modulus | 82 GPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bulk modulus | 170 GPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Poisson ratio | 0.29 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mohs hardness | 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Vickers hardness | 608 MPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Brinell hardness | 200–1180 MPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAS Number | 7439-89-6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| History | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Discovery | before 5000 BC | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symbol | «Fe»: from Latin ferrum | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Main isotopes of iron
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| references |
Iron ( or ) is a chemical element with symbol Fe (from Latin: ferrum) and atomic number 26. It is a metal that belongs to the first transition series and group 8 of the periodic table. It is, by mass, the most common element on Earth, just ahead of oxygen (32.1% and 30.1%, respectively), forming much of Earth’s outer and inner core. It is the fourth most common element in the Earth’s crust, being mainly deposited by meteorites in its metallic state, with its ores also being found there.
Extracting usable metal from iron ores requires kilns or furnaces capable of reaching 1,500 °C (2,730 °F) or higher, about 500 °C (932 °F) higher than that required to smelt copper. Humans started to master that process in Eurasia during the 2nd millennium BCE and the use of iron tools and weapons began to displace copper alloys—in some regions, only around 1200 BCE. That event is considered the transition from the Bronze Age to the Iron Age. In the modern world, iron alloys, such as steel, stainless steel, cast iron and special steels, are by far the most common industrial metals, due to their mechanical properties and low cost. The iron and steel industry is thus very important economically, and iron is the cheapest metal, with a price of a few dollars per kilogram or pound.
Pristine and smooth pure iron surfaces are a mirror-like silvery-gray. Iron reacts readily with oxygen and water to produce brown-to-black hydrated iron oxides, commonly known as rust. Unlike the oxides of some other metals that form passivating layers, rust occupies more volume than the metal and thus flakes off, exposing more fresh surfaces for corrosion. High-purity irons (e.g. electrolytic iron) are more resistant to corrosion.
The body of an adult human contains about 4 grams (0.005% body weight) of iron, mostly in hemoglobin and myoglobin. These two proteins play essential roles in vertebrate metabolism, respectively oxygen transport by blood and oxygen storage in muscles. To maintain the necessary levels, human iron metabolism requires a minimum of iron in the diet. Iron is also the metal at the active site of many important redox enzymes dealing with cellular respiration and oxidation and reduction in plants and animals.[5]
Chemically, the most common oxidation states of iron are iron(II) and iron(III). Iron shares many properties of other transition metals, including the other group 8 elements, ruthenium and osmium. Iron forms compounds in a wide range of oxidation states, −2 to +7. Iron also forms many coordination compounds; some of them, such as ferrocene, ferrioxalate, and Prussian blue have substantial industrial, medical, or research applications.
Characteristics
Allotropes
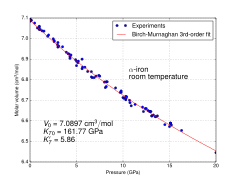
Molar volume vs. pressure for α iron at room temperature
At least four allotropes of iron (differing atom arrangements in the solid) are known, conventionally denoted α, γ, δ, and ε.

The first three forms are observed at ordinary pressures. As molten iron cools past its freezing point of 1538 °C, it crystallizes into its δ allotrope, which has a body-centered cubic (bcc) crystal structure. As it cools further to 1394 °C, it changes to its γ-iron allotrope, a face-centered cubic (fcc) crystal structure, or austenite. At 912 °C and below, the crystal structure again becomes the bcc α-iron allotrope.[6]
The physical properties of iron at very high pressures and temperatures have also been studied extensively,[7][8] because of their relevance to theories about the cores of the Earth and other planets. Above approximately 10 GPa and temperatures of a few hundred kelvin or less, α-iron changes into another hexagonal close-packed (hcp) structure, which is also known as ε-iron. The higher-temperature γ-phase also changes into ε-iron, but does so at higher pressure.
Some controversial experimental evidence exists for a stable β phase at pressures above 50 GPa and temperatures of at least 1500 K. It is supposed to have an orthorhombic or a double hcp structure.[9] (Confusingly, the term «β-iron» is sometimes also used to refer to α-iron above its Curie point, when it changes from being ferromagnetic to paramagnetic, even though its crystal structure has not changed.[6])
The inner core of the Earth is generally presumed to consist of an iron-nickel alloy with ε (or β) structure.[10]
Melting and boiling points
The melting and boiling points of iron, along with its enthalpy of atomization, are lower than those of the earlier 3d elements from scandium to chromium, showing the lessened contribution of the 3d electrons to metallic bonding as they are attracted more and more into the inert core by the nucleus;[11] however, they are higher than the values for the previous element manganese because that element has a half-filled 3d sub-shell and consequently its d-electrons are not easily delocalized. This same trend appears for ruthenium but not osmium.[12]
The melting point of iron is experimentally well defined for pressures less than 50 GPa. For greater pressures, published data (as of 2007) still varies by tens of gigapascals and over a thousand kelvin.[13]
Magnetic properties
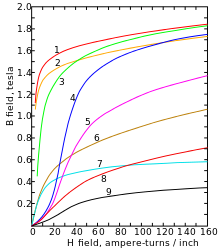
Magnetization curves of 9 ferromagnetic materials, showing saturation. 1. Sheet steel, 2. Silicon steel, 3. Cast steel, 4. Tungsten steel, 5. Magnet steel, 6. Cast iron, 7. Nickel, 8. Cobalt, 9. Magnetite[14]
Below its Curie point of 770 °C (1,420 °F; 1,040 K), α-iron changes from paramagnetic to ferromagnetic: the spins of the two unpaired electrons in each atom generally align with the spins of its neighbors, creating an overall magnetic field.[15] This happens because the orbitals of those two electrons (dz2 and dx2 −. y2) do not point toward neighboring atoms in the lattice, and therefore are not involved in metallic bonding.[6]
In the absence of an external source of magnetic field, the atoms get spontaneously partitioned into magnetic domains, about 10 micrometers across,[16] such that the atoms in each domain have parallel spins, but some domains have other orientations. Thus a macroscopic piece of iron will have a nearly zero overall magnetic field.
Application of an external magnetic field causes the domains that are magnetized in the same general direction to grow at the expense of adjacent ones that point in other directions, reinforcing the external field. This effect is exploited in devices that need to channel magnetic fields to fulfill design function, such as electrical transformers, magnetic recording heads, and electric motors. Impurities, lattice defects, or grain and particle boundaries can «pin» the domains in the new positions, so that the effect persists even after the external field is removed – thus turning the iron object into a (permanent) magnet.[15]
Similar behavior is exhibited by some iron compounds, such as the ferrites including the mineral magnetite, a crystalline form of the mixed iron(II,III) oxide Fe3O4 (although the atomic-scale mechanism, ferrimagnetism, is somewhat different). Pieces of magnetite with natural permanent magnetization (lodestones) provided the earliest compasses for navigation. Particles of magnetite were extensively used in magnetic recording media such as core memories, magnetic tapes, floppies, and disks, until they were replaced by cobalt-based materials.
Isotopes
Iron has four stable isotopes: 54Fe (5.845% of natural iron), 56Fe (91.754%), 57Fe (2.119%) and 58Fe (0.282%). 24 artificial isotopes have also been created. Of these stable isotopes, only 57Fe has a nuclear spin (−1⁄2). The nuclide 54Fe theoretically can undergo double electron capture to 54Cr, but the process has never been observed and only a lower limit on the half-life of 3.1×1022 years has been established.[17]
60Fe is an extinct radionuclide of long half-life (2.6 million years).[18] It is not found on Earth, but its ultimate decay product is its granddaughter, the stable nuclide 60Ni.[17] Much of the past work on isotopic composition of iron has focused on the nucleosynthesis of 60Fe through studies of meteorites and ore formation. In the last decade, advances in mass spectrometry have allowed the detection and quantification of minute, naturally occurring variations in the ratios of the stable isotopes of iron. Much of this work is driven by the Earth and planetary science communities, although applications to biological and industrial systems are emerging.[19]
In phases of the meteorites Semarkona and Chervony Kut, a correlation between the concentration of 60Ni, the granddaughter of 60Fe, and the abundance of the stable iron isotopes provided evidence for the existence of 60Fe at the time of formation of the Solar System. Possibly the energy released by the decay of 60Fe, along with that released by 26Al, contributed to the remelting and differentiation of asteroids after their formation 4.6 billion years ago. The abundance of 60Ni present in extraterrestrial material may bring further insight into the origin and early history of the Solar System.[20]
The most abundant iron isotope 56Fe is of particular interest to nuclear scientists because it represents the most common endpoint of nucleosynthesis.[21] Since 56Ni (14 alpha particles) is easily produced from lighter nuclei in the alpha process in nuclear reactions in supernovae (see silicon burning process), it is the endpoint of fusion chains inside extremely massive stars, since addition of another alpha particle, resulting in 60Zn, requires a great deal more energy. This 56Ni, which has a half-life of about 6 days, is created in quantity in these stars, but soon decays by two successive positron emissions within supernova decay products in the supernova remnant gas cloud, first to radioactive 56Co, and then to stable 56Fe. As such, iron is the most abundant element in the core of red giants, and is the most abundant metal in iron meteorites and in the dense metal cores of planets such as Earth.[22] It is also very common in the universe, relative to other stable metals of approximately the same atomic weight.[22][23] Iron is the sixth most abundant element in the universe, and the most common refractory element.[24]
Although a further tiny energy gain could be extracted by synthesizing 62Ni, which has a marginally higher binding energy than 56Fe, conditions in stars are unsuitable for this process. Element production in supernovas greatly favor iron over nickel, and in any case, 56Fe still has a lower mass per nucleon than 62Ni due to its higher fraction of lighter protons.[25] Hence, elements heavier than iron require a supernova for their formation, involving rapid neutron capture by starting 56Fe nuclei.[22]
In the far future of the universe, assuming that proton decay does not occur, cold fusion occurring via quantum tunnelling would cause the light nuclei in ordinary matter to fuse into 56Fe nuclei. Fission and alpha-particle emission would then make heavy nuclei decay into iron, converting all stellar-mass objects to cold spheres of pure iron.[26]
Origin and occurrence in nature
Cosmogenesis
Iron’s abundance in rocky planets like Earth is due to its abundant production during the runaway fusion and explosion of type Ia supernovae, which scatters the iron into space.[27][28]
Metallic iron

A polished and chemically etched piece of an iron meteorite, believed to be similar in composition to the Earth’s metallic core, showing individual crystals of the iron-nickel alloy (Widmanstatten pattern)
Metallic or native iron is rarely found on the surface of the Earth because it tends to oxidize. However, both the Earth’s inner and outer core, that account for 35% of the mass of the whole Earth, are believed to consist largely of an iron alloy, possibly with nickel. Electric currents in the liquid outer core are believed to be the origin of the Earth’s magnetic field. The other terrestrial planets (Mercury, Venus, and Mars) as well as the Moon are believed to have a metallic core consisting mostly of iron. The M-type asteroids are also believed to be partly or mostly made of metallic iron alloy.
The rare iron meteorites are the main form of natural metallic iron on the Earth’s surface. Items made of cold-worked meteoritic iron have been found in various archaeological sites dating from a time when iron smelting had not yet been developed; and the Inuit in Greenland have been reported to use iron from the Cape York meteorite for tools and hunting weapons.[29] About 1 in 20 meteorites consist of the unique iron-nickel minerals taenite (35–80% iron) and kamacite (90–95% iron).[30] Native iron is also rarely found in basalts that have formed from magmas that have come into contact with carbon-rich sedimentary rocks, which have reduced the oxygen fugacity sufficiently for iron to crystallize. This is known as Telluric iron and is described from a few localities, such as Disko Island in West Greenland, Yakutia in Russia and Bühl in Germany.[31]
Mantle minerals
Ferropericlase (Mg,Fe)O, a solid solution of periclase (MgO) and wüstite (FeO), makes up about 20% of the volume of the lower mantle of the Earth, which makes it the second most abundant mineral phase in that region after silicate perovskite (Mg,Fe)SiO3; it also is the major host for iron in the lower mantle.[32] At the bottom of the transition zone of the mantle, the reaction γ-(Mg,Fe)2[SiO4] ↔ (Mg,Fe)[SiO3] + (Mg,Fe)O transforms γ-olivine into a mixture of silicate perovskite and ferropericlase and vice versa. In the literature, this mineral phase of the lower mantle is also often called magnesiowüstite.[33] Silicate perovskite may form up to 93% of the lower mantle,[34] and the magnesium iron form, (Mg,Fe)SiO3, is considered to be the most abundant mineral in the Earth, making up 38% of its volume.[35]
Earth’s crust

While iron is the most abundant element on Earth, most of this iron is concentrated in the inner and outer cores.[36][37] The fraction of iron that is in Earth’s crust only amounts to about 5% of the overall mass of the crust and is thus only the fourth most abundant element in that layer (after oxygen, silicon, and aluminium).[38]
Most of the iron in the crust is combined with various other elements to form many iron minerals. An important class is the iron oxide minerals such as hematite (Fe2O3), magnetite (Fe3O4), and siderite (FeCO3), which are the major ores of iron. Many igneous rocks also contain the sulfide minerals pyrrhotite and pentlandite.[39][40] During weathering, iron tends to leach from sulfide deposits as the sulfate and from silicate deposits as the bicarbonate. Both of these are oxidized in aqueous solution and precipitate in even mildly elevated pH as iron(III) oxide.[41]

Banded iron formation in McKinley Park, Minnesota.
Large deposits of iron are banded iron formations, a type of rock consisting of repeated thin layers of iron oxides alternating with bands of iron-poor shale and chert. The banded iron formations were laid down in the time between 3,700 million years ago and 1,800 million years ago.[42][43]
Materials containing finely ground iron(III) oxides or oxide-hydroxides, such as ochre, have been used as yellow, red, and brown pigments since pre-historical times. They contribute as well to the color of various rocks and clays, including entire geological formations like the Painted Hills in Oregon and the Buntsandstein («colored sandstone», British Bunter).[44] Through Eisensandstein (a jurassic ‘iron sandstone’, e.g. from Donzdorf in Germany)[45] and Bath stone in the UK, iron compounds are responsible for the yellowish color of many historical buildings and sculptures.[46] The proverbial red color of the surface of Mars is derived from an iron oxide-rich regolith.[47]
Significant amounts of iron occur in the iron sulfide mineral pyrite (FeS2), but it is difficult to extract iron from it and it is therefore not exploited.[48] In fact, iron is so common that production generally focuses only on ores with very high quantities of it.[49]
According to the International Resource Panel’s Metal Stocks in Society report, the global stock of iron in use in society is 2,200 kg per capita. More-developed countries differ in this respect from less-developed countries (7,000–14,000 vs 2,000 kg per capita).[50]
Oceans
Ocean science demonstrated the role of the iron in the ancient seas in both marine biota and climate.[51]
Chemistry and compounds
| Oxidation state | Representative compound |
|---|---|
| −2 (d10) | Disodium tetracarbonylferrate (Collman’s reagent) |
| −1 (d9) | Fe 2(CO)2− 8 |
| 0 (d8) | Iron pentacarbonyl |
| 1 (d7) | Cyclopentadienyliron dicarbonyl dimer («Fp2«) |
| 2 (d6) | Ferrous sulfate, ferrocene |
| 3 (d5) | Ferric chloride, ferrocenium tetrafluoroborate |
| 4 (d4) | Fe(diars) 2Cl2+ 2, Ferryl tetrafluoroborate |
| 5 (d3) | FeO3− 4 |
| 6 (d2) | Potassium ferrate |
| 7 (d1) | [FeO4]– (matrix isolation, 4K) |
Iron shows the characteristic chemical properties of the transition metals, namely the ability to form variable oxidation states differing by steps of one and a very large coordination and organometallic chemistry: indeed, it was the discovery of an iron compound, ferrocene, that revolutionalized the latter field in the 1950s.[52] Iron is sometimes considered as a prototype for the entire block of transition metals, due to its abundance and the immense role it has played in the technological progress of humanity.[53] Its 26 electrons are arranged in the configuration [Ar]3d64s2, of which the 3d and 4s electrons are relatively close in energy, and thus a number of electrons can be ionized.[12]
Iron forms compounds mainly in the oxidation states +2 (iron(II), «ferrous») and +3 (iron(III), «ferric»). Iron also occurs in higher oxidation states, e.g., the purple potassium ferrate (K2FeO4), which contains iron in its +6 oxidation state. The anion [FeO4]– with iron in its +7 oxidation state, along with an iron(V)-peroxo isomer, has been detected by infrared spectroscopy at 4 K after cocondensation of laser-ablated Fe atoms with a mixture of O2/Ar.[54] Iron(IV) is a common intermediate in many biochemical oxidation reactions.[55][56] Numerous organoiron compounds contain formal oxidation states of +1, 0, −1, or even −2. The oxidation states and other bonding properties are often assessed using the technique of Mössbauer spectroscopy.[57] Many mixed valence compounds contain both iron(II) and iron(III) centers, such as magnetite and Prussian blue (Fe4(Fe[CN]6)3).[56] The latter is used as the traditional «blue» in blueprints.[58]
Iron is the first of the transition metals that cannot reach its group oxidation state of +8, although its heavier congeners ruthenium and osmium can, with ruthenium having more difficulty than osmium.[6] Ruthenium exhibits an aqueous cationic chemistry in its low oxidation states similar to that of iron, but osmium does not, favoring high oxidation states in which it forms anionic complexes.[6] In the second half of the 3d transition series, vertical similarities down the groups compete with the horizontal similarities of iron with its neighbors cobalt and nickel in the periodic table, which are also ferromagnetic at room temperature and share similar chemistry. As such, iron, cobalt, and nickel are sometimes grouped together as the iron triad.[53]
Unlike many other metals, iron does not form amalgams with mercury. As a result, mercury is traded in standardized 76 pound flasks (34 kg) made of iron.[59]
Iron is by far the most reactive element in its group; it is pyrophoric when finely divided and dissolves easily in dilute acids, giving Fe2+. However, it does not react with concentrated nitric acid and other oxidizing acids due to the formation of an impervious oxide layer, which can nevertheless react with hydrochloric acid.[6] High-purity iron, called electrolytic iron, is considered to be resistant to rust, due to its oxide layer.
Binary compounds
Oxides and sulfides

Ferrous or iron(II) oxide,
FeO

Ferric or iron(III) oxide
Fe2O3

Ferrosoferric or iron(II,III) oxide
Fe3O4
Iron forms various oxide and hydroxide compounds; the most common are iron(II,III) oxide (Fe3O4), and iron(III) oxide (Fe2O3). Iron(II) oxide also exists, though it is unstable at room temperature. Despite their names, they are actually all non-stoichiometric compounds whose compositions may vary.[60] These oxides are the principal ores for the production of iron (see bloomery and blast furnace). They are also used in the production of ferrites, useful magnetic storage media in computers, and pigments. The best known sulfide is iron pyrite (FeS2), also known as fool’s gold owing to its golden luster.[56] It is not an iron(IV) compound, but is actually an iron(II) polysulfide containing Fe2+ and S2−
2 ions in a distorted sodium chloride structure.[60]
Halides

Hydrated iron(III) chloride (ferric chloride)
The binary ferrous and ferric halides are well-known. The ferrous halides typically arise from treating iron metal with the corresponding hydrohalic acid to give the corresponding hydrated salts.[56]
- Fe + 2 HX → FeX2 + H2 (X = F, Cl, Br, I)
Iron reacts with fluorine, chlorine, and bromine to give the corresponding ferric halides, ferric chloride being the most common.[61]
- 2 Fe + 3 X2 → 2 FeX3 (X = F, Cl, Br)
Ferric iodide is an exception, being thermodynamically unstable due to the oxidizing power of Fe3+ and the high reducing power of I−:[61]
- 2 I− + 2 Fe3+ → I2 + 2 Fe2+ (E0 = +0.23 V)
Ferric iodide, a black solid, is not stable in ordinary conditions, but can be prepared through the reaction of iron pentacarbonyl with iodine and carbon monoxide in the presence of hexane and light at the temperature of −20 °C, with oxygen and water excluded.[61]Complexes of ferric iodide with some soft bases are known to be stable compounds.[62][63]
Solution chemistry

Comparison of colors of solutions of ferrate (left) and permanganate (right)
The standard reduction potentials in acidic aqueous solution for some common iron ions are given below:[6]
| [Fe(H2O)6]2+ + 2 e− | ⇌ Fe | E0 = −0.447 V |
| [Fe(H2O)6]3+ + e− | ⇌ [Fe(H2O)6]2+ | E0 = +0.77 V |
| FeO2− 4 + 8 H3O+ + 3 e− | ⇌ [Fe(H2O)6]3+ + 6 H2O | E0 = +2.20 V |
The red-purple tetrahedral ferrate(VI) anion is such a strong oxidizing agent that it oxidizes ammonia to nitrogen (N2) and water to oxygen[61]
- 4 FeO2−
4 + 34 H
2O → 4 [Fe(H2O)6]3+ + 20 OH−
+ 3 O2
The pale-violet hexaquo complex [Fe(H2O)6]3+ is an acid such that above above pH 0 it is fully hydrolyzed:[64]
| [Fe(H2O)6]3+ | ⇌ [Fe(H2O)5(OH)]2+ + H+ | K = 10−3.05 mol dm−3 |
| [Fe(H2O)5(OH)]2+ | ⇌ [Fe(H2O)4(OH)2]+ + H+ | K = 10−3.26 mol dm−3 |
| 2[Fe(H2O)6]3+ | ⇌ [Fe(H2O)4(OH)]4+2 + 2H+ + 2H2O | K = 10−2.91 mol dm−3 |

As pH rises above 0 the above yellow hydrolyzed species form and as it rises above 2–3, reddish-brown hydrous iron(III) oxide precipitates out of solution. Although Fe3+ has a d5 configuration, its absorption spectrum is not like that of Mn2+ with its weak, spin-forbidden d–d bands, because Fe3+ has higher positive charge and is more polarizing, lowering the energy of its ligand-to-metal charge transfer absorptions. Thus, all the above complexes are rather strongly colored, with the single exception of the hexaquo ion – and even that has a spectrum dominated by charge transfer in the near ultraviolet region.[64] On the other hand, the pale green iron(II) hexaquo ion [Fe(H2O)6]2+ does not undergo appreciable hydrolysis. Carbon dioxide is not evolved when carbonate anions are added, which instead results in white iron(II) carbonate being precipitated out. In excess carbon dioxide this forms the slightly soluble bicarbonate, which occurs commonly in groundwater, but it oxidises quickly in air to form iron(III) oxide that accounts for the brown deposits present in a sizeable number of streams.[65]
Coordination compounds
Due to its electronic structure, iron has a very large coordination and organometallic chemistry.

Many coordination compounds of iron are known. A typical six-coordinate anion is hexachloroferrate(III), [FeCl6]3−, found in the mixed salt tetrakis(methylammonium) hexachloroferrate(III) chloride.[66][67] Complexes with multiple bidentate ligands have geometric isomers. For example, the trans-chlorohydridobis(bis-1,2-(diphenylphosphino)ethane)iron(II) complex is used as a starting material for compounds with the Fe(dppe)2 moiety.[68][69] The ferrioxalate ion with three oxalate ligands (shown at right) displays helical chirality with its two non-superposable geometries labelled Λ (lambda) for the left-handed screw axis and Δ (delta) for the right-handed screw axis, in line with IUPAC conventions.[64] Potassium ferrioxalate is used in chemical actinometry and along with its sodium salt undergoes photoreduction applied in old-style photographic processes. The dihydrate of iron(II) oxalate has a polymeric structure with co-planar oxalate ions bridging between iron centres with the water of crystallisation located forming the caps of each octahedron, as illustrated below.[70]

Crystal structure of iron(II) oxalate dihydrate, showing iron (gray), oxygen (red), carbon (black), and hydrogen (white) atoms.

Blood-red positive thiocyanate test for iron(III)
Iron(III) complexes are quite similar to those of chromium(III) with the exception of iron(III)’s preference for O-donor instead of N-donor ligands. The latter tend to be rather more unstable than iron(II) complexes and often dissociate in water. Many Fe–O complexes show intense colors and are used as tests for phenols or enols. For example, in the ferric chloride test, used to determine the presence of phenols, iron(III) chloride reacts with a phenol to form a deep violet complex:[64]
- 3 ArOH + FeCl3 → Fe(OAr)3 + 3 HCl (Ar = aryl)
Among the halide and pseudohalide complexes, fluoro complexes of iron(III) are the most stable, with the colorless [FeF5(H2O)]2− being the most stable in aqueous solution. Chloro complexes are less stable and favor tetrahedral coordination as in [FeCl4]−; [FeBr4]− and [FeI4]− are reduced easily to iron(II). Thiocyanate is a common test for the presence of iron(III) as it forms the blood-red [Fe(SCN)(H2O)5]2+. Like manganese(II), most iron(III) complexes are high-spin, the exceptions being those with ligands that are high in the spectrochemical series such as cyanide. An example of a low-spin iron(III) complex is [Fe(CN)6]3−. Iron shows a great variety of electronic spin states, including every possible spin quantum number value for a d-block element from 0 (diamagnetic) to 5⁄2 (5 unpaired electrons). This value is always half the number of unpaired electrons. Complexes with zero to two unpaired electrons are considered low-spin and those with four or five are considered high-spin.[60]
Iron(II) complexes are less stable than iron(III) complexes but the preference for O-donor ligands is less marked, so that for example [Fe(NH3)6]2+ is known while [Fe(NH3)6]3+ is not. They have a tendency to be oxidized to iron(III) but this can be moderated by low pH and the specific ligands used.[65]
Organometallic compounds

Organoiron chemistry is the study of organometallic compounds of iron, where carbon atoms are covalently bound to the metal atom. They are many and varied, including cyanide complexes, carbonyl complexes, sandwich and half-sandwich compounds.
![]()
Prussian blue or «ferric ferrocyanide», Fe4[Fe(CN)6]3, is an old and well-known iron-cyanide complex, extensively used as pigment and in several other applications. Its formation can be used as a simple wet chemistry test to distinguish between aqueous solutions of Fe2+ and Fe3+ as they react (respectively) with potassium ferricyanide and potassium ferrocyanide to form Prussian blue.[56]
Another old example of an organoiron compound is iron pentacarbonyl, Fe(CO)5, in which a neutral iron atom is bound to the carbon atoms of five carbon monoxide molecules. The compound can be used to make carbonyl iron powder, a highly reactive form of metallic iron. Thermolysis of iron pentacarbonyl gives triiron dodecacarbonyl, Fe3(CO)12, a complex with a cluster of three iron atoms at its core. Collman’s reagent, disodium tetracarbonylferrate, is a useful reagent for organic chemistry; it contains iron in the −2 oxidation state. Cyclopentadienyliron dicarbonyl dimer contains iron in the rare +1 oxidation state.[71]


Structural formula of ferrocene and a powdered sample
A landmark in this field was the discovery in 1951 of the remarkably stable sandwich compound ferrocene Fe(C5H5)2, by Pauson and Kealy[72] and independently by Miller and colleagues,[73] whose surprising molecular structure was determined only a year later by Woodward and Wilkinson[74] and Fischer.[75]
Ferrocene is still one of the most important tools and models in this class.[76]
Iron-centered organometallic species are used as catalysts. The Knölker complex, for example, is a transfer hydrogenation catalyst for ketones.[77]
Industrial uses
The iron compounds produced on the largest scale in industry are iron(II) sulfate (FeSO4·7H2O) and iron(III) chloride (FeCl3). The former is one of the most readily available sources of iron(II), but is less stable to aerial oxidation than Mohr’s salt ((NH4)2Fe(SO4)2·6H2O). Iron(II) compounds tend to be oxidized to iron(III) compounds in the air.[56]
History
Development of iron metallurgy
Iron is one of the elements undoubtedly known to the ancient world.[78] It has been worked, or wrought, for millennia. However, iron artefacts of great age are much rarer than objects made of gold or silver due to the ease with which iron corrodes.[79] The technology developed slowly, and even after the discovery of smelting it took many centuries for iron to replace bronze as the metal of choice for tools and weapons.
Meteoritic iron

Beads made from meteoric iron in 3500 BC or earlier were found in Gerzeh, Egypt by G.A. Wainwright.[80] The beads contain 7.5% nickel, which is a signature of meteoric origin since iron found in the Earth’s crust generally has only minuscule nickel impurities.
Meteoric iron was highly regarded due to its origin in the heavens and was often used to forge weapons and tools.[80] For example, a dagger made of meteoric iron was found in the tomb of Tutankhamun, containing similar proportions of iron, cobalt, and nickel to a meteorite discovered in the area, deposited by an ancient meteor shower.[81][82][83] Items that were likely made of iron by Egyptians date from 3000 to 2500 BC.[79]
Meteoritic iron is comparably soft and ductile and easily cold forged but may get brittle when heated because of the nickel content.[84]
Wrought iron

The symbol for Mars has been used since antiquity to represent iron.

The iron pillar of Delhi is an example of the iron extraction and processing methodologies of early India.
The first iron production started in the Middle Bronze Age, but it took several centuries before iron displaced bronze. Samples of smelted iron from Asmar, Mesopotamia and Tall Chagar Bazaar in northern Syria were made sometime between 3000 and 2700 BC.[85] The Hittites established an empire in north-central Anatolia around 1600 BC. They appear to be the first to understand the production of iron from its ores and regard it highly in their society.[86] The Hittites began to smelt iron between 1500 and 1200 BC and the practice spread to the rest of the Near East after their empire fell in 1180 BC.[85] The subsequent period is called the Iron Age.
Artifacts of smelted iron are found in India dating from 1800 to 1200 BC,[87] and in the Levant from about 1500 BC (suggesting smelting in Anatolia or the Caucasus).[88][89] Alleged references (compare history of metallurgy in South Asia) to iron in the Indian Vedas have been used for claims of a very early usage of iron in India respectively to date the texts as such. The rigveda term ayas (metal) refers to copper, while iron which is called as śyāma ayas, literally «black copper», first is mentioned in the post-rigvedic Atharvaveda.[90]
Some archaeological evidence suggests iron was smelted in Zimbabwe and southeast Africa as early as the eighth century BC.[91] Iron working was introduced to Greece in the late 11th century BC, from which it spread quickly throughout Europe.[92]

Iron sickle from Ancient Greece.
The spread of ironworking in Central and Western Europe is associated with Celtic expansion. According to Pliny the Elder, iron use was common in the Roman era.[80] In the lands of what is now considered China, iron appears approximately 700–500 BC.[93] Iron smelting may have been introduced into China through Central Asia.[94] The earliest evidence of the use of a blast furnace in China dates to the 1st century AD,[95] and cupola furnaces were used as early as the Warring States period (403–221 BC).[96] Usage of the blast and cupola furnace remained widespread during the Tang and Song dynasties.[97]
During the Industrial Revolution in Britain, Henry Cort began refining iron from pig iron to wrought iron (or bar iron) using innovative production systems. In 1783 he patented the puddling process for refining iron ore. It was later improved by others, including Joseph Hall.[98]
Cast iron
Cast iron was first produced in China during 5th century BC,[99] but was hardly in Europe until the medieval period.[100][101] The earliest cast iron artifacts were discovered by archaeologists in what is now modern Luhe County, Jiangsu in China. Cast iron was used in ancient China for warfare, agriculture, and architecture.[102] During the medieval period, means were found in Europe of producing wrought iron from cast iron (in this context known as pig iron) using finery forges. For all these processes, charcoal was required as fuel.[103]

Medieval blast furnaces were about 10 feet (3.0 m) tall and made of fireproof brick; forced air was usually provided by hand-operated bellows.[101] Modern blast furnaces have grown much bigger, with hearths fourteen meters in diameter that allow them to produce thousands of tons of iron each day, but essentially operate in much the same way as they did during medieval times.[103]
In 1709, Abraham Darby I established a coke-fired blast furnace to produce cast iron, replacing charcoal, although continuing to use blast furnaces. The ensuing availability of inexpensive iron was one of the factors leading to the Industrial Revolution. Toward the end of the 18th century, cast iron began to replace wrought iron for certain purposes, because it was cheaper. Carbon content in iron was not implicated as the reason for the differences in properties of wrought iron, cast iron, and steel until the 18th century.[85]
Since iron was becoming cheaper and more plentiful, it also became a major structural material following the building of the innovative first iron bridge in 1778. This bridge still stands today as a monument to the role iron played in the Industrial Revolution. Following this, iron was used in rails, boats, ships, aqueducts, and buildings, as well as in iron cylinders in steam engines.[103] Railways have been central to the formation of modernity and ideas of progress[104] and various languages refer to railways as iron road (e.g. French chemin de fer, German Eisenbahn, Turkish demiryolu, Russian железная дорога, Chinese, Japanese, and Korean 鐵道, Vietnamese đường sắt).
Steel
Steel (with smaller carbon content than pig iron but more than wrought iron) was first produced in antiquity by using a bloomery. Blacksmiths in Luristan in western Persia were making good steel by 1000 BC.[85] Then improved versions, Wootz steel by India and Damascus steel were developed around 300 BC and AD 500 respectively. These methods were specialized, and so steel did not become a major commodity until the 1850s.[105]
New methods of producing it by carburizing bars of iron in the cementation process were devised in the 17th century. In the Industrial Revolution, new methods of producing bar iron without charcoal were devised and these were later applied to produce steel. In the late 1850s, Henry Bessemer invented a new steelmaking process, involving blowing air through molten pig iron, to produce mild steel. This made steel much more economical, thereby leading to wrought iron no longer being produced in large quantities.[106]
Foundations of modern chemistry
In 1774, Antoine Lavoisier used the reaction of water steam with metallic iron inside an incandescent iron tube to produce hydrogen in his experiments leading to the demonstration of the conservation of mass, which was instrumental in changing chemistry from a qualitative science to a quantitative one.[107]
Symbolic role

«Gold gab ich für Eisen» – «I gave gold for iron». German-American brooch from WWI.
Iron plays a certain role in mythology and has found various usage as a metaphor and in folklore. The Greek poet Hesiod’s Works and Days (lines 109–201) lists different ages of man named after metals like gold, silver, bronze and iron to account for successive ages of humanity.[108] The Iron Age was closely related with Rome, and in Ovid’s Metamorphoses
The Virtues, in despair, quit the earth; and the depravity of man becomes universal and complete. Hard steel succeeded then.
An example of the importance of iron’s symbolic role may be found in the German Campaign of 1813. Frederick William III commissioned then the first Iron Cross as military decoration. Berlin iron jewellery reached its peak production between 1813 and 1815, when the Prussian royal family urged citizens to donate gold and silver jewellery for military funding. The inscription Gold gab ich für Eisen (I gave gold for iron) was used as well in later war efforts.[109]
Production of metallic iron


Iron furnace in Columbus, Ohio, 1922
Laboratory routes
For a few limited purposes when it is needed, pure iron is produced in the laboratory in small quantities by reducing the pure oxide or hydroxide with hydrogen, or forming iron pentacarbonyl and heating it to 250 °C so that it decomposes to form pure iron powder.[41] Another method is electrolysis of ferrous chloride onto an iron cathode.[110]
Main industrial route
| Country | Iron ore | Pig iron | Direct iron | Steel |
|---|---|---|---|---|
| 1,114.9 | 549.4 | 573.6 | ||
| 393.9 | 4.4 | 5.2 | ||
| 305.0 | 25.1 | 0.011 | 26.5 | |
| 66.9 | 87.5 | |||
| 257.4 | 38.2 | 23.4 | 63.5 | |
| 92.1 | 43.9 | 4.7 | 60.0 | |
| 65.8 | 25.7 | 29.9 | ||
| 0.1 | 27.3 | 48.6 | ||
| 0.4 | 20.1 | 0.38 | 32.7 | |
| World | 1,594.9 | 914.0 | 64.5 | 1,232.4 |
Nowadays, the industrial production of iron or steel consists of two main stages. In the first stage, iron ore is reduced with coke in a blast furnace, and the molten metal is separated from gross impurities such as silicate minerals. This stage yields an alloy—pig iron—that contains relatively large amounts of carbon. In the second stage, the amount of carbon in the pig iron is lowered by oxidation to yield wrought iron, steel, or cast iron.[112] Other metals can be added at this stage to form alloy steels.

17th century Chinese illustration of workers at a blast furnace, making wrought iron from pig iron[113]

How iron was extracted in the 19th century
Blast furnace processing
The blast furnace is loaded with iron ores, usually hematite Fe2O3 or magnetite Fe3O4, along with coke (coal that has been separately baked to remove volatile components) and flux (limestone or dolomite). «Blasts» of air pre-heated to 900 °C (sometimes with oxygen enrichment) is blown through the mixture, in sufficient amount to turn the carbon into carbon monoxide:[112]

This reaction raises the temperature to about 2000 °C. The carbon monoxide reduces the iron ore to metallic iron[112]

Some iron in the high-temperature lower region of the furnace reacts directly with the coke:[112]

The flux removes silicaceous minerals in the ore, which would otherwise clog the furnace: The heat of the furnace decomposes the carbonates to calcium oxide, which reacts with any excess silica to form a slag composed of calcium silicate CaSiO3 or other products. At the furnace’s temperature, the metal and the slag are both molten. They collect at the bottom as two immiscible liquid layers (with the slag on top), that are then easily separated.[112] The slag can be used as a material in road construction or to improve mineral-poor soils for agriculture.[101]
Steelmaking thus remains one of the largest industrial contributors of CO2 emissions in the world.[114]

This heap of iron ore pellets will be used in steel production.
Steelmaking

A pot of molten iron being used to make steel
The pig iron produced by the blast furnace process contains up to 4–5% carbon (by mass), with small amounts of other impurities like sulfur, magnesium, phosphorus, and manganese. This high level of carbon makes it relatively weak and brittle. Reducing the amount of carbon to 0.002–2.1% produces steel, which may be up to 1000 times harder than pure iron. A great variety of steel articles can then be made by cold working, hot rolling, forging, machining, etc. Removing the impurities from pig iron, but leaving 2–4% carbon, results in cast iron, which is cast by foundries into articles such as stoves, pipes, radiators, lamp-posts, and rails.[112]
Steel products often undergo various heat treatments after they are forged to shape. Annealing consists of heating them to 700–800 °C for several hours and then gradual cooling. It makes the steel softer and more workable.[115]
Direct iron reduction
Owing to environmental concerns, alternative methods of processing iron have been developed. «Direct iron reduction» reduces iron ore to a ferrous lump called «sponge» iron or «direct» iron that is suitable for steelmaking.[101] Two main reactions comprise the direct reduction process:
Natural gas is partially oxidized (with heat and a catalyst):[101]

Iron ore is then treated with these gases in a furnace, producing solid sponge iron:[101]

Silica is removed by adding a limestone flux as described above.[101]
Thermite process
Ignition of a mixture of aluminium powder and iron oxide yields metallic iron via the thermite reaction:

Alternatively pig iron may be made into steel (with up to about 2% carbon) or wrought iron (commercially pure iron). Various processes have been used for this, including finery forges, puddling furnaces, Bessemer converters, open hearth furnaces, basic oxygen furnaces, and electric arc furnaces. In all cases, the objective is to oxidize some or all of the carbon, together with other impurities. On the other hand, other metals may be added to make alloy steels.[103]
Applications
As structural material
Iron is the most widely used of all the metals, accounting for over 90% of worldwide metal production. Its low cost and high strength often make it the material of choice to withstand stress or transmit forces, such as the construction of machinery and machine tools, rails, automobiles, ship hulls, concrete reinforcing bars, and the load-carrying framework of buildings. Since pure iron is quite soft, it is most commonly combined with alloying elements to make steel.[116]
Mechanical properties
| Material | TS (MPa) | BH (Brinell) |
|---|---|---|
| Iron whiskers | 11000 | |
| Ausformed (hardened) steel | 2930 | 850–1200 |
| Martensitic steel | 2070 | 600 |
| Bainitic steel | 1380 | 400 |
| Pearlitic steel | 1200 | 350 |
| Cold-worked iron | 690 | 200 |
| Small-grain iron | 340 | 100 |
| Carbon-containing iron | 140 | 40 |
| Pure, single-crystal iron | 10 | 3 |
The mechanical properties of iron and its alloys are extremely relevant to their structural applications. Those properties can be evaluated in various ways, including the Brinell test, the Rockwell test and the Vickers hardness test.
The properties of pure iron are often used to calibrate measurements or to compare tests.[118][119] However, the mechanical properties of iron are significantly affected by the sample’s purity: pure, single crystals of iron are actually softer than aluminium,[117] and the purest industrially produced iron (99.99%) has a hardness of 20–30 Brinell.[120] The pure iron (99.9%~99.999%), especially called electrolytic iron, is industrially produced by electrolytic refining.
An increase in the carbon content will cause a significant increase in the hardness and tensile strength of iron. Maximum hardness of 65 Rc is achieved with a 0.6% carbon content, although the alloy has low tensile strength.[121] Because of the softness of iron, it is much easier to work with than its heavier congeners ruthenium and osmium.[12]

Iron-carbon phase diagram
Types of steels and alloys
α-Iron is a fairly soft metal that can dissolve only a small concentration of carbon (no more than 0.021% by mass at 910 °C).[122] Austenite (γ-iron) is similarly soft and metallic but can dissolve considerably more carbon (as much as 2.04% by mass at 1146 °C). This form of iron is used in the type of stainless steel used for making cutlery, and hospital and food-service equipment.[16]
Commercially available iron is classified based on purity and the abundance of additives. Pig iron has 3.5–4.5% carbon[123] and contains varying amounts of contaminants such as sulfur, silicon and phosphorus. Pig iron is not a saleable product, but rather an intermediate step in the production of cast iron and steel. The reduction of contaminants in pig iron that negatively affect material properties, such as sulfur and phosphorus, yields cast iron containing 2–4% carbon, 1–6% silicon, and small amounts of manganese.[112] Pig iron has a melting point in the range of 1420–1470 K, which is lower than either of its two main components, and makes it the first product to be melted when carbon and iron are heated together.[6] Its mechanical properties vary greatly and depend on the form the carbon takes in the alloy.[12]
«White» cast irons contain their carbon in the form of cementite, or iron carbide (Fe3C).[12] This hard, brittle compound dominates the mechanical properties of white cast irons, rendering them hard, but unresistant to shock. The broken surface of a white cast iron is full of fine facets of the broken iron carbide, a very pale, silvery, shiny material, hence the appellation. Cooling a mixture of iron with 0.8% carbon slowly below 723 °C to room temperature results in separate, alternating layers of cementite and α-iron, which is soft and malleable and is called pearlite for its appearance. Rapid cooling, on the other hand, does not allow time for this separation and creates hard and brittle martensite. The steel can then be tempered by reheating to a temperature in between, changing the proportions of pearlite and martensite. The end product below 0.8% carbon content is a pearlite-αFe mixture, and that above 0.8% carbon content is a pearlite-cementite mixture.[12]
In gray iron the carbon exists as separate, fine flakes of graphite, and also renders the material brittle due to the sharp edged flakes of graphite that produce stress concentration sites within the material.[124] A newer variant of gray iron, referred to as ductile iron, is specially treated with trace amounts of magnesium to alter the shape of graphite to spheroids, or nodules, reducing the stress concentrations and vastly increasing the toughness and strength of the material.[124]
Wrought iron contains less than 0.25% carbon but large amounts of slag that give it a fibrous characteristic.[123] It is a tough, malleable product, but not as fusible as pig iron. If honed to an edge, it loses it quickly. Wrought iron is characterized by the presence of fine fibers of slag entrapped within the metal. Wrought iron is more corrosion resistant than steel. It has been almost completely replaced by mild steel for traditional «wrought iron» products and blacksmithing.
Mild steel corrodes more readily than wrought iron, but is cheaper and more widely available. Carbon steel contains 2.0% carbon or less,[125] with small amounts of manganese, sulfur, phosphorus, and silicon. Alloy steels contain varying amounts of carbon as well as other metals, such as chromium, vanadium, molybdenum, nickel, tungsten, etc. Their alloy content raises their cost, and so they are usually only employed for specialist uses. One common alloy steel, though, is stainless steel. Recent developments in ferrous metallurgy have produced a growing range of microalloyed steels, also termed ‘HSLA’ or high-strength, low alloy steels, containing tiny additions to produce high strengths and often spectacular toughness at minimal cost.[125][126][127]
Alloys with high purity elemental makeups (such as alloys of electrolytic iron) have specifically enhanced properties such as ductility, tensile strength, toughness, fatigue strength, heat resistance, and corrosion resistance.
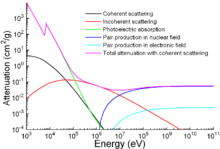
Apart from traditional applications, iron is also used for protection from ionizing radiation. Although it is lighter than another traditional protection material, lead, it is much stronger mechanically. The attenuation of radiation as a function of energy is shown in the graph.[128]
The main disadvantage of iron and steel is that pure iron, and most of its alloys, suffer badly from rust if not protected in some way, a cost amounting to over 1% of the world’s economy.[129] Painting, galvanization, passivation, plastic coating and bluing are all used to protect iron from rust by excluding water and oxygen or by cathodic protection. The mechanism of the rusting of iron is as follows:[129]
- Cathode: 3 O2 + 6 H2O + 12 e− → 12 OH−
- Anode: 4 Fe → 4 Fe2+ + 8 e−; 4 Fe2+ → 4 Fe3+ + 4 e−
- Overall: 4 Fe + 3 O2 + 6 H2O → 4 Fe3+ + 12 OH− → 4 Fe(OH)3 or 4 FeO(OH) + 4 H2O
The electrolyte is usually iron(II) sulfate in urban areas (formed when atmospheric sulfur dioxide attacks iron), and salt particles in the atmosphere in seaside areas.[129]
Catalysts and reagents
Because Fe is inexpensive and nontoxic, much effort has been devoted to the development of Fe-based catalysts and reagents. Iron is however less common as a catalyst in commercial processes than more expensive metals.[130] In biology, Fe-containing enzymes are pervasive.[131]
Iron catalysts are traditionally used in the Haber–Bosch process for the production of ammonia and the Fischer–Tropsch process for conversion of carbon monoxide to hydrocarbons for fuels and lubricants.[132] Powdered iron in an acidic medium is used in the Bechamp reduction, the conversion of nitrobenzene to aniline.[133]
Iron compounds
Iron(III) oxide mixed with aluminium powder can be ignited to create a thermite reaction, used in welding large iron parts (like rails) and purifying ores. Iron(III) oxide and oxyhydroxide are used as reddish and ocher pigments.
Iron(III) chloride finds use in water purification and sewage treatment, in the dyeing of cloth, as a coloring agent in paints, as an additive in animal feed, and as an etchant for copper in the manufacture of printed circuit boards.[134] It can also be dissolved in alcohol to form tincture of iron, which is used as a medicine to stop bleeding in canaries.[135]
Iron(II) sulfate is used as a precursor to other iron compounds. It is also used to reduce chromate in cement. It is used to fortify foods and treat iron deficiency anemia. Iron(III) sulfate is used in settling minute sewage particles in tank water. Iron(II) chloride is used as a reducing flocculating agent, in the formation of iron complexes and magnetic iron oxides, and as a reducing agent in organic synthesis.[134]
Sodium nitroprusside is a drug used as a vasodilator. It is on the World Health Organization’s List of Essential Medicines.[136]
Biological and pathological role
Iron is required for life.[5][137][138] The iron–sulfur clusters are pervasive and include nitrogenase, the enzymes responsible for biological nitrogen fixation. Iron-containing proteins participate in transport, storage and used of oxygen.[5] Iron proteins are involved in electron transfer.[139]
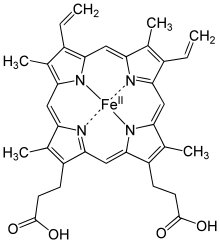
Simplified structure of Heme b; in the protein additional ligand(s) are attached to Fe.
Examples of iron-containing proteins in higher organisms include hemoglobin, cytochrome (see high-valent iron), and catalase.[5][140] The average adult human contains about 0.005% body weight of iron, or about four grams, of which three quarters is in hemoglobin – a level that remains constant despite only about one milligram of iron being absorbed each day,[139] because the human body recycles its hemoglobin for the iron content.[141]
Microbial growth may be assisted by oxidation of iron(II) or by reduction of iron (III).[142]
Biochemistry
Iron acquisition poses a problem for aerobic organisms because ferric iron is poorly soluble near neutral pH. Thus, these organisms have developed means to absorb iron as complexes, sometimes taking up ferrous iron before oxidising it back to ferric iron.[5] In particular, bacteria have evolved very high-affinity sequestering agents called siderophores.[143][144][145]
After uptake in human cells, iron storage is precisely regulated.[5][146] A major component of this regulation is the protein transferrin, which binds iron ions absorbed from the duodenum and carries it in the blood to cells.[5][147] Transferrin contains Fe3+ in the middle of a distorted octahedron, bonded to one nitrogen, three oxygens and a chelating carbonate anion that traps the Fe3+ ion: it has such a high stability constant that it is very effective at taking up Fe3+ ions even from the most stable complexes. At the bone marrow, transferrin is reduced from Fe3+ and Fe2+ and stored as ferritin to be incorporated into hemoglobin.[139]
The most commonly known and studied bioinorganic iron compounds (biological iron molecules) are the heme proteins: examples are hemoglobin, myoglobin, and cytochrome P450.[5] These compounds participate in transporting gases, building enzymes, and transferring electrons.[139] Metalloproteins are a group of proteins with metal ion cofactors. Some examples of iron metalloproteins are ferritin and rubredoxin.[139] Many enzymes vital to life contain iron, such as catalase,[148] lipoxygenases,[149] and IRE-BP.[150]
Hemoglobin is an oxygen carrier that occurs in red blood cells and contributes their color, transporting oxygen in the arteries from the lungs to the muscles where it is transferred to myoglobin, which stores it until it is needed for the metabolic oxidation of glucose, generating energy.[5] Here the hemoglobin binds to carbon dioxide, produced when glucose is oxidized, which is transported through the veins by hemoglobin (predominantly as bicarbonate anions) back to the lungs where it is exhaled.[139] In hemoglobin, the iron is in one of four heme groups and has six possible coordination sites; four are occupied by nitrogen atoms in a porphyrin ring, the fifth by an imidazole nitrogen in a histidine residue of one of the protein chains attached to the heme group, and the sixth is reserved for the oxygen molecule it can reversibly bind to.[139] When hemoglobin is not attached to oxygen (and is then called deoxyhemoglobin), the Fe2+ ion at the center of the heme group (in the hydrophobic protein interior) is in a high-spin configuration. It is thus too large to fit inside the porphyrin ring, which bends instead into a dome with the Fe2+ ion about 55 picometers above it. In this configuration, the sixth coordination site reserved for the oxygen is blocked by another histidine residue.[139]
When deoxyhemoglobin picks up an oxygen molecule, this histidine residue moves away and returns once the oxygen is securely attached to form a hydrogen bond with it. This results in the Fe2+ ion switching to a low-spin configuration, resulting in a 20% decrease in ionic radius so that now it can fit into the porphyrin ring, which becomes planar.[139] (Additionally, this hydrogen bonding results in the tilting of the oxygen molecule, resulting in a Fe–O–O bond angle of around 120° that avoids the formation of Fe–O–Fe or Fe–O2–Fe bridges that would lead to electron transfer, the oxidation of Fe2+ to Fe3+, and the destruction of hemoglobin.) This results in a movement of all the protein chains that leads to the other subunits of hemoglobin changing shape to a form with larger oxygen affinity. Thus, when deoxyhemoglobin takes up oxygen, its affinity for more oxygen increases, and vice versa.[139] Myoglobin, on the other hand, contains only one heme group and hence this cooperative effect cannot occur. Thus, while hemoglobin is almost saturated with oxygen in the high partial pressures of oxygen found in the lungs, its affinity for oxygen is much lower than that of myoglobin, which oxygenates even at low partial pressures of oxygen found in muscle tissue.[139] As described by the Bohr effect (named after Christian Bohr, the father of Niels Bohr), the oxygen affinity of hemoglobin diminishes in the presence of carbon dioxide.[139]

Carbon monoxide and phosphorus trifluoride are poisonous to humans because they bind to hemoglobin similarly to oxygen, but with much more strength, so that oxygen can no longer be transported throughout the body. Hemoglobin bound to carbon monoxide is known as carboxyhemoglobin. This effect also plays a minor role in the toxicity of cyanide, but there the major effect is by far its interference with the proper functioning of the electron transport protein cytochrome a.[139] The cytochrome proteins also involve heme groups and are involved in the metabolic oxidation of glucose by oxygen. The sixth coordination site is then occupied by either another imidazole nitrogen or a methionine sulfur, so that these proteins are largely inert to oxygen – with the exception of cytochrome a, which bonds directly to oxygen and thus is very easily poisoned by cyanide.[139] Here, the electron transfer takes place as the iron remains in low spin but changes between the +2 and +3 oxidation states. Since the reduction potential of each step is slightly greater than the previous one, the energy is released step-by-step and can thus be stored in adenosine triphosphate. Cytochrome a is slightly distinct, as it occurs at the mitochondrial membrane, binds directly to oxygen, and transports protons as well as electrons, as follows:[139]
- 4 Cytc2+ + O2 + 8H+
inside → 4 Cytc3+ + 2 H2O + 4H+
outside
Although the heme proteins are the most important class of iron-containing proteins, the iron–sulfur proteins are also very important, being involved in electron transfer, which is possible since iron can exist stably in either the +2 or +3 oxidation states. These have one, two, four, or eight iron atoms that are each approximately tetrahedrally coordinated to four sulfur atoms; because of this tetrahedral coordination, they always have high-spin iron. The simplest of such compounds is rubredoxin, which has only one iron atom coordinated to four sulfur atoms from cysteine residues in the surrounding peptide chains. Another important class of iron–sulfur proteins is the ferredoxins, which have multiple iron atoms. Transferrin does not belong to either of these classes.[139]
The ability of sea mussels to maintain their grip on rocks in the ocean is facilitated by their use of organometallic iron-based bonds in their protein-rich cuticles. Based on synthetic replicas, the presence of iron in these structures increased elastic modulus 770 times, tensile strength 58 times, and toughness 92 times. The amount of stress required to permanently damage them increased 76 times.[152]
Nutrition
Diet
Iron is pervasive, but particularly rich sources of dietary iron include red meat, oysters, beans, poultry, fish, leaf vegetables, watercress, tofu, and blackstrap molasses.[5] Bread and breakfast cereals are sometimes specifically fortified with iron.[5][153]
Iron provided by dietary supplements is often found as iron(II) fumarate, although iron(II) sulfate is cheaper and is absorbed equally well.[134] Elemental iron, or reduced iron, despite being absorbed at only one-third to two-thirds the efficiency (relative to iron sulfate),[154] is often added to foods such as breakfast cereals or enriched wheat flour. Iron is most available to the body when chelated to amino acids[155] and is also available for use as a common iron supplement. Glycine, the least expensive amino acid, is most often used to produce iron glycinate supplements.[156]
Dietary recommendations
The U.S. Institute of Medicine (IOM) updated Estimated Average Requirements (EARs) and Recommended Dietary Allowances (RDAs) for iron in 2001.[5] The current EAR for iron for women ages 14–18 is 7.9 mg/day, 8.1 for ages 19–50 and 5.0 thereafter (post menopause). For men the EAR is 6.0 mg/day for ages 19 and up. The RDA is 15.0 mg/day for women ages 15–18, 18.0 for 19–50 and 8.0 thereafter. For men, 8.0 mg/day for ages 19 and up. RDAs are higher than EARs so as to identify amounts that will cover people with higher than average requirements. RDA for pregnancy is 27 mg/day and, for lactation, 9 mg/day.[5] For children ages 1–3 years 7 mg/day, 10 for ages 4–8 and 8 for ages 9–13. As for safety, the IOM also sets Tolerable upper intake levels (ULs) for vitamins and minerals when evidence is sufficient. In the case of iron the UL is set at 45 mg/day. Collectively the EARs, RDAs and ULs are referred to as Dietary Reference Intakes.[157]
The European Food Safety Authority (EFSA) refers to the collective set of information as Dietary Reference Values, with Population Reference Intake (PRI) instead of RDA, and Average Requirement instead of EAR. AI and UL defined the same as in United States. For women the PRI is 13 mg/day ages 15–17 years, 16 mg/day for women ages 18 and up who are premenopausal and 11 mg/day postmenopausal. For pregnancy and lactation, 16 mg/day. For men the PRI is 11 mg/day ages 15 and older. For children ages 1 to 14 the PRI increases from 7 to 11 mg/day. The PRIs are higher than the U.S. RDAs, with the exception of pregnancy.[158] The EFSA reviewed the same safety question did not establish a UL.[159]
Infants may require iron supplements if they are bottle-fed cow’s milk.[160] Frequent blood donors are at risk of low iron levels and are often advised to supplement their iron intake.[161]
For U.S. food and dietary supplement labeling purposes the amount in a serving is expressed as a percent of Daily Value (%DV). For iron labeling purposes 100% of the Daily Value was 18 mg, and as of May 27, 2016 remained unchanged at 18 mg.[162][163] A table of the old and new adult daily values is provided at Reference Daily Intake.
Deficiency
Iron deficiency is the most common nutritional deficiency in the world.[5][164][165][166] When loss of iron is not adequately compensated by adequate dietary iron intake, a state of latent iron deficiency occurs, which over time leads to iron-deficiency anemia if left untreated, which is characterised by an insufficient number of red blood cells and an insufficient amount of hemoglobin.[167] Children, pre-menopausal women (women of child-bearing age), and people with poor diet are most susceptible to the disease. Most cases of iron-deficiency anemia are mild, but if not treated can cause problems like fast or irregular heartbeat, complications during pregnancy, and delayed growth in infants and children.[168]
Excess
Iron uptake is tightly regulated by the human body, which has no regulated physiological means of excreting iron. Only small amounts of iron are lost daily due to mucosal and skin epithelial cell sloughing, so control of iron levels is primarily accomplished by regulating uptake.[169] Regulation of iron uptake is impaired in some people as a result of a genetic defect that maps to the HLA-H gene region on chromosome 6 and leads to abnormally low levels of hepcidin, a key regulator of the entry of iron into the circulatory system in mammals.[170] In these people, excessive iron intake can result in iron overload disorders, known medically as hemochromatosis.[5] Many people have an undiagnosed genetic susceptibility to iron overload, and are not aware of a family history of the problem. For this reason, people should not take iron supplements unless they suffer from iron deficiency and have consulted a doctor. Hemochromatosis is estimated to be the cause of 0.3 to 0.8% of all metabolic diseases of Caucasians.[171]
Overdoses of ingested iron can cause excessive levels of free iron in the blood. High blood levels of free ferrous iron react with peroxides to produce highly reactive free radicals that can damage DNA, proteins, lipids, and other cellular components. Iron toxicity occurs when the cell contains free iron, which generally occurs when iron levels exceed the availability of transferrin to bind the iron. Damage to the cells of the gastrointestinal tract can also prevent them from regulating iron absorption, leading to further increases in blood levels. Iron typically damages cells in the heart, liver and elsewhere, causing adverse effects that include coma, metabolic acidosis, shock, liver failure, coagulopathy, long-term organ damage, and even death.[172] Humans experience iron toxicity when the iron exceeds 20 milligrams for every kilogram of body mass; 60 milligrams per kilogram is considered a lethal dose.[173] Overconsumption of iron, often the result of children eating large quantities of ferrous sulfate tablets intended for adult consumption, is one of the most common toxicological causes of death in children under six.[173] The Dietary Reference Intake (DRI) sets the Tolerable Upper Intake Level (UL) for adults at 45 mg/day. For children under fourteen years old the UL is 40 mg/day.[174]
The medical management of iron toxicity is complicated, and can include use of a specific chelating agent called deferoxamine to bind and expel excess iron from the body.[172][175][176]
ADHD
Some research has suggested that low thalamic iron levels may play a role in the pathophysiology of ADHD.[177] Some researchers have found that iron supplementation can be effective especially in the inattentive subtype of the disorder.[178] One study also showed that iron may be able to decrease the risk of cardiovascular events during treatment with ADHD drugs.[179]
Some researchers in the 2000s suggested a link between low levels of iron in the blood and ADHD. A 2012 study found no such correlation.[180]
Cancer
The role of iron in cancer defense can be described as a «double-edged sword» because of its pervasive presence in non-pathological processes.[181] People having chemotherapy may develop iron deficiency and anemia, for which intravenous iron therapy is used to restore iron levels.[182] Iron overload, which may occur from high consumption of red meat,[5] may initiate tumor growth and increase susceptibility to cancer onset,[182] particularly for colorectal cancer.[5]
Marine systems
Iron plays an essential role in marine systems and can act as a limiting nutrient for planktonic activity.[183] Because of this, too much of a decrease in iron may lead to a decrease in growth rates in phytoplanktonic organisms such as diatoms.[184] Iron can also be oxidized by marine microbes under conditions that are high in iron and low in oxygen.[185]
Iron can enter marine systems through adjoining rivers and directly from the atmosphere. Once iron enters the ocean, it can be distributed throughout the water column through ocean mixing and through recycling on the cellular level.[186] In the arctic, sea ice plays a major role in the store and distribution of iron in the ocean, depleting oceanic iron as it freezes in the winter and releasing it back into the water when thawing occurs in the summer.[187] The iron cycle can fluctuate the forms of iron from aqueous to particle forms altering the availability of iron to primary producers.[188] Increased light and warmth increases the amount of iron that is in forms that are usable by primary producers.[189]
See also
- El Mutún in Bolivia, where 10% of the world’s accessible iron ore is located
- Iron and steel industry
- Iron cycle
- Iron nanoparticle
- Iron–platinum nanoparticle
- Iron fertilization – proposed fertilization of oceans to stimulate phytoplankton growth
- Iron-oxidizing bacteria
- List of countries by iron production
- Pelletising – process of creation of iron ore pellets
- Rustproof iron
- Steel
References
- ^ «Standard Atomic Weights: Iron». CIAAW. 1993.
- ^ Ram, R. S.; Bernath, P. F. (2003). «Fourier transform emission spectroscopy of the g4Δ–a4Δ system of FeCl». Journal of Molecular Spectroscopy. 221 (2): 261. Bibcode:2003JMoSp.221..261R. doi:10.1016/S0022-2852(03)00225-X.
- ^ Demazeau, G.; Buffat, B.; Pouchard, M.; Hagenmuller, P. (1982). «Recent developments in the field of high oxidation states of transition elements in oxides stabilization of six-coordinated Iron(V)». Zeitschrift für anorganische und allgemeine Chemie. 491: 60–66. doi:10.1002/zaac.19824910109.
- ^ Lu, J.; Jian, J.; Huang, W.; Lin, H.; Li, J; Zhou, M. (2016). «Experimental and theoretical identification of the Fe(VII) oxidation state in FeO4−«. Physical Chemistry Chemical Physics. 18 (45): 31125–31131. Bibcode:2016PCCP…1831125L. doi:10.1039/C6CP06753K. PMID 27812577.
- ^ a b c d e f g h i j k l m n o p q «Iron». Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis, Oregon. April 2016. Retrieved 6 March 2018.
- ^ a b c d e f g h Greenwood & Earnshaw 1997, pp. 1075–79.
- ^ Hirose, K., Tateno, S. (2010). «The Structure of Iron in Earth’s Inner Core». Science. American Association for the Advancement of Science. 330 (6002): 359–361. Bibcode:2010Sci…330..359T. doi:10.1126/science.1194662. PMID 20947762. S2CID 206528628.
- ^ Chamati, Gaminchev (2014). «Dynamic stability of Fe under high pressure». Journal of Physics. IOP Publishing. 558 (1): 012013. Bibcode:2014JPhCS.558a2013G. doi:10.1088/1742-6596/558/1/012013.
- ^ Boehler, Reinhard (2000). «High-pressure experiments and the phase diagram of lower mantle and core materials». Reviews of Geophysics. American Geophysical Union. 38 (2): 221–45. Bibcode:2000RvGeo..38..221B. doi:10.1029/1998RG000053. S2CID 33458168.
- ^ Stixrude, Lars; Wasserman, Evgeny; Cohen, Ronald E. (10 November 1997). «Composition and temperature of Earth’s inner core». Journal of Geophysical Research: Solid Earth. 102 (B11): 24729–39. Bibcode:1997JGR…10224729S. doi:10.1029/97JB02125.
- ^ Greenwood & Earnshaw 1997, p. 1116.
- ^ a b c d e f Greenwood & Earnshaw 1997, pp. 1074–75.
- ^ Boehler, Reinhard; Ross, M. (2007). «Properties of Rocks and Minerals_High-Pressure Melting». Mineral Physics. Treatise on Geophysics. Vol. 2. Elsevier. pp. 527–41. doi:10.1016/B978-044452748-6.00047-X. ISBN 9780444527486.
- ^ Steinmetz, Charles (1917). «fig. 42». Theory and Calculation of Electric Circuits. McGraw-Hill.
- ^ a b Cullity; C. D. Graham (2008). Introduction to Magnetic Materials, 2nd. New York: Wiley–IEEE. p. 116. ISBN 978-0-471-47741-9.
- ^ a b Bramfitt, B.L.; Benscoter, Arlan O. (2002). «The Iron Carbon Phase Diagram». Metallographer’s guide: practice and procedures for irons and steels. ASM International. pp. 24–28. ISBN 978-0-87170-748-2.
- ^ a b Audi, Georges; Bersillon, Olivier; Blachot, Jean; Wapstra, Aaldert Hendrik (2003), «The NUBASE evaluation of nuclear and decay properties», Nuclear Physics A, 729: 3–128, Bibcode:2003NuPhA.729….3A, doi:10.1016/j.nuclphysa.2003.11.001
- ^ Rugel, G.; Faestermann, T.; Knie, K.; Korschinek, G.; Poutivtsev, M.; Schumann, D.; Kivel, N.; Günther-Leopold, I.; Weinreich, R.; Wohlmuther, M. (2009). «New Measurement of the 60Fe Half-Life». Physical Review Letters. 103 (7): 072502. Bibcode:2009PhRvL.103g2502R. doi:10.1103/PhysRevLett.103.072502. PMID 19792637.
- ^ Dauphas, N.; Rouxel, O. (2006). «Mass spectrometry and natural variations of iron isotopes» (PDF). Mass Spectrometry Reviews. 25 (4): 515–50. Bibcode:2006MSRv…25..515D. doi:10.1002/mas.20078. PMID 16463281. Archived from the original (PDF) on 10 June 2010.
- ^ Mostefaoui, S.; Lugmair, G.W.; Hoppe, P.; El Goresy, A. (2004). «Evidence for live 60Fe in meteorites». New Astronomy Reviews. 48 (1–4): 155–59. Bibcode:2004NewAR..48..155M. doi:10.1016/j.newar.2003.11.022.
- ^ Fewell, M. P. (1995). «The atomic nuclide with the highest mean binding energy». American Journal of Physics. 63 (7): 653. Bibcode:1995AmJPh..63..653F. doi:10.1119/1.17828.
- ^ a b c Greenwood & Earnshaw 1997, p. 12.
- ^ Woosley, S.; Janka, T. (2006). «The physics of core collapse supernovae». Nature Physics. 1 (3): 147–54. arXiv:astro-ph/0601261. Bibcode:2005NatPh…1..147W. doi:10.1038/nphys172. S2CID 118974639.
- ^ McDonald, I.; Sloan, G. C.; Zijlstra, A. A.; Matsunaga, N.; Matsuura, M.; Kraemer, K. E.; Bernard-Salas, J.; Markwick, A. J. (2010). «Rusty Old Stars: A Source of the Missing Interstellar Iron?». The Astrophysical Journal Letters. 717 (2): L92–L97. arXiv:1005.3489. Bibcode:2010ApJ…717L..92M. doi:10.1088/2041-8205/717/2/L92. S2CID 14437704.
- ^ Bautista, Manuel A.; Pradhan, Anil K. (1995). «Iron and Nickel Abundances in H~II Regions and Supernova Remnants». Bulletin of the American Astronomical Society. 27: 865. Bibcode:1995AAS…186.3707B.
- ^ Dyson, Freeman J. (1979). «Time without end: Physics and biology in an open universe». Reviews of Modern Physics. 51 (3): 447–60. Bibcode:1979RvMP…51..447D. doi:10.1103/RevModPhys.51.447.
- ^ Aron, Jacob. «Supernova space bullets could have seeded Earth’s iron core». New Scientist. Retrieved 2 October 2020.
- ^ Croswell, Ken. «Iron in the Fire: The Little-Star Supernovae That Could». Scientific American. Retrieved 3 January 2021.
- ^ Buchwald, V F (1992). «On the Use of Iron by the Eskimos in Greenland». Materials Characterization. 29 (2): 139–176. doi:10.1016/1044-5803(92)90112-U.
- ^ Emiliani, Cesare (1992). Planet earth: cosmology, geology, and the evolution of life and environment. Cambridge University Press. p. 152. Bibcode:1992pecg.book…..E. ISBN 978-0-521-40949-0.
- ^ Pernet-Fisher, J.; Day, J.M.D.; Howarth, G.H.; Ryabov, V.V.; Taylor, L.A. (2017). «Atmospheric outgassing and native-iron formation during carbonaceous sediment–basalt melt interactions». Earth and Planetary Science Letters. 460: 201–212. Bibcode:2017E&PSL.460..201P. doi:10.1016/j.epsl.2016.12.022.
- ^ Stark, Anne M. (20 September 2007) Researchers locate mantle’s spin transition zone, leading to clues about earth’s structure. Lawrence Livermore National Laboratory
- ^ Ferropericlase. Mindat.org
- ^ Murakami, M.; Ohishi Y.; Hirao N.; Hirose K. (2012). «A perovskitic lower mantle inferred from high-pressure, high-temperature sound velocity data». Nature. 485 (7396): 90–94. Bibcode:2012Natur.485…90M. doi:10.1038/nature11004. PMID 22552097. S2CID 4387193.
- ^ Sharp, T. (27 November 2014). «Bridgmanite – named at last». Science. 346 (6213): 1057–58. Bibcode:2014Sci…346.1057S. doi:10.1126/science.1261887. PMID 25430755. S2CID 206563252.
- ^ Kong, L. T.; Li, J. F.; Shi, Q. W.; Huang, H. J.; Zhao, K. (6 March 2012). «Dynamical stability of iron under high-temperature and high-pressure conditions». EPL. 97 (5): 56004p1–56004p5. Bibcode:2012EL…..9756004K. doi:10.1209/0295-5075/97/56004. S2CID 121861429.
- ^ Gaminchev, K. G.; Chamati, H. (3 December 2014). «Dynamic stability of Fe under high pressure». J. Phys. 558 (1): 012013(1–7). Bibcode:2014JPhCS.558a2013G. doi:10.1088/1742-6596/558/1/012013.
- ^ Morgan, John W. & Anders, Edward (1980). «Chemical composition of Earth, Venus, and Mercury». Proc. Natl. Acad. Sci. 77 (12): 6973–77. Bibcode:1980PNAS…77.6973M. doi:10.1073/pnas.77.12.6973. PMC 350422. PMID 16592930.
- ^ «Pyrrhotite». Mindat.org. Retrieved 7 July 2009.
- ^ Klein, Cornelis and Cornelius S. Hurlbut, Jr. (1985) Manual of Mineralogy, Wiley, 20th ed, pp. 278–79 ISBN 0-471-80580-7
- ^ a b Greenwood & Earnshaw 1997, p. 1071.
- ^ Lyons, T. W.; Reinhard, C. T. (2009). «Early Earth: Oxygen for heavy-metal fans». Nature. 461 (7261): 179–181. Bibcode:2009Natur.461..179L. doi:10.1038/461179a. PMID 19741692. S2CID 205049360.
- ^ Cloud, P. (1973). «Paleoecological Significance of the Banded Iron-Formation». Economic Geology. 68 (7): 1135–43. doi:10.2113/gsecongeo.68.7.1135.
- ^ Dickinson, Robert E. (1964). Germany: A regional and economic geography (2nd ed.). London: Methuen.
- ^ Naturwerksteine in Baden-Württemberg. Landesamt für Geologie, Rohstoffe und Bergbau, Baden-Württemberg
- ^ «Tales From The Riverbank». Minerva Stone Conservation. Archived from the original on 28 September 2015. Retrieved 22 September 2015.
- ^ Klingelhöfer, G.; Morris, R. V.; Souza, P. A.; Rodionov, D.; Schröder, C. (2007). «Two earth years of Mössbauer studies of the surface of Mars with MIMOS II». Hyperfine Interactions. 170 (1–3): 169–77. Bibcode:2006HyInt.170..169K. doi:10.1007/s10751-007-9508-5. S2CID 98227499.
- ^ Winderlich, R.; Peter, W. (1954). Lehrbuch der Chemie für Höhere Lehranstalten : Einheitsausgabe für Unter- und Oberstufe (in German). Wiesbaden. p. 75. ISBN 978-3-663-04370-6. OCLC 913701506.
- ^ Bertau, Martin (2013). Industrielle Anorganische Chemie (in German). Weinheim: Wiley-VCH. p. 696. ISBN 978-3-527-64956-3. OCLC 855858511.
- ^ Metal Stocks in Society: Scientific synthesis, 2010, International Resource Panel, UNEP
- ^ Stoll, Heather (17 February 2020). «30 years of the iron hypothesis of ice ages». Nature. Springer Science and Business Media LLC. 578 (7795): 370–371. doi:10.1038/d41586-020-00393-x. ISSN 0028-0836.
- ^ Greenwood & Earnshaw 1997, p. 905.
- ^ a b Greenwood & Earnshaw 1997, p. 1070.
- ^ Lu, Jun-Bo; Jian, Jiwen; Huang, Wei; Lin, Hailu; Li, Jun; Zhou, Mingfei (16 November 2016). «Experimental and theoretical identification of the Fe(VII) oxidation state in FeO4−«. Phys. Chem. Chem. Phys. 18 (45): 31125–31131. Bibcode:2016PCCP…1831125L. doi:10.1039/c6cp06753k. PMID 27812577.
- ^ Nam, Wonwoo (2007). «High-Valent Iron(IV)–Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions» (PDF). Accounts of Chemical Research. 40 (7): 522–531. doi:10.1021/ar700027f. PMID 17469792. Archived from the original (PDF) on 15 June 2021. Retrieved 22 February 2022.
- ^ a b c d e f Holleman, Arnold F.; Wiberg, Egon; Wiberg, Nils (1985). «Iron». Lehrbuch der Anorganischen Chemie (in German) (91–100 ed.). Walter de Gruyter. pp. 1125–46. ISBN 3-11-007511-3.
- ^ Reiff, William Michael; Long, Gary J. (1984). «Mössbauer Spectroscopy and the Coordination Chemistry of Iron». Mössbauer spectroscopy applied to inorganic chemistry. Springer. pp. 245–83. ISBN 978-0-306-41647-7.
- ^ Ware, Mike (1999). «An introduction in monochrome». Cyanotype: the history, science and art of photographic printing in Prussian blue. NMSI Trading Ltd. pp. 11–19. ISBN 978-1-900747-07-3.
- ^ Gmelin, Leopold (1852). «Mercury and Iron». Hand-book of chemistry. Vol. 6. Cavendish Society. pp. 128–29.
- ^ a b c Greenwood & Earnshaw 1997, p. 1079.
- ^ a b c d Greenwood & Earnshaw 1997, pp. 1082–84.
- ^ Siegfried Pohl, Ulrich Bierbach, Wolfgang Saak; «FeI3SC(NMe2)2, a Neutral Thiourea Complex of Iron(III) Iodide», Angewandte Chemie International Edition in English (1989) 28 (6), 776-777. https://doi.org/10.1002/anie.198907761
- ^ Nicholas A. Barnes, Stephen M.Godfrey, Nicholas Ho, Charles A.McAuliffe, Robin G.Pritchard; «Facile synthesis of a rare example of an iron(III) iodide complex, [FeI3(AsMe3)2], from the reaction of Me3AsI2 with unactivated iron powder», Polyhedron (2013) 55, 67-72. https://doi.org/10.1016/j.poly.2013.02.066
- ^ a b c d Greenwood & Earnshaw 1997, pp. 1088–91.
- ^ a b Greenwood & Earnshaw 1997, pp. 1091–97.
- ^ Clausen, C.A.; Good, M.L. (1968). «Stabilization of the hexachloroferrate(III) anion by the methylammonium cation». Inorganic Chemistry. 7 (12): 2662–63. doi:10.1021/ic50070a047.
- ^ James, B.D.; Bakalova, M.; Lieseganga, J.; Reiff, W.M.; Hockless, D.C.R.; Skelton, B.W.; White, A.H. (1996). «The hexachloroferrate(III) anion stabilized in hydrogen bonded packing arrangements. A comparison of the X-ray crystal structures and low temperature magnetism of tetrakis(methylammonium) hexachloroferrate(III) chloride (I) and tetrakis(hexamethylenediammonium) hexachloroferrate(III) tetrachloroferrate(III) tetrachloride (II)«. Inorganica Chimica Acta. 247 (2): 169–74. doi:10.1016/0020-1693(95)04955-X.
- ^ Giannoccaro, P.; Sacco, A. (1977). Bis[ethylenebis(diphenylphosphine)]-Hydridoiron Complexes. Inorg. Synth. Inorganic Syntheses. Vol. 17. pp. 69–72. doi:10.1002/9780470132487.ch19. ISBN 978-0-470-13248-7.
- ^ Lee, J.; Jung, G.; Lee, S.W. (1998). «Structure of trans-chlorohydridobis(diphenylphosphinoethane)iron(II)». Bull. Korean Chem. Soc. 19 (2): 267–69. doi:10.1007/BF02698412. S2CID 35665289.
- ^ Echigo, Takuya; Kimata, Mitsuyoshi (2008). «Single-crystal X-ray diffraction and spectroscopic studies on humboldtine and lindbergite: weak Jahn–Teller effect of Fe2+ ion». Phys. Chem. Minerals. 35 (8): 467–75. Bibcode:2008PCM….35..467E. doi:10.1007/s00269-008-0241-7. S2CID 98739882.
- ^ Greenwood, Norman N.; Earnshaw, Alan (1984). Chemistry of the Elements. Oxford: Pergamon Press. pp. 1282–86. ISBN 978-0-08-022057-4..
- ^ Kealy, T.J.; Pauson, P.L. (1951). «A New Type of Organo-Iron Compound». Nature. 168 (4285): 1039–40. Bibcode:1951Natur.168.1039K. doi:10.1038/1681039b0. S2CID 4181383.
- ^ Miller, S. A.; Tebboth, J. A.; Tremaine, J. F. (1952). «114. Dicyclopentadienyliron». J. Chem. Soc.: 632–635. doi:10.1039/JR9520000632.
- ^ Wilkinson, G.; Rosenblum, M.; Whiting, M. C.; Woodward, R. B. (1952). «The Structure of Iron Bis-Cyclopentadienyl». J. Am. Chem. Soc. 74 (8): 2125–2126. doi:10.1021/ja01128a527.
- ^ Okuda, Jun (28 December 2016). «Ferrocene – 65 Years After». European Journal of Inorganic Chemistry. 2017 (2): 217–219. doi:10.1002/ejic.201601323. ISSN 1434-1948.
- ^ Greenwood & Earnshaw 1997, p. 1104.
- ^ Bullock, R.M. (11 September 2007). «An Iron Catalyst for Ketone Hydrogenations under Mild Conditions». Angew. Chem. Int. Ed. 46 (39): 7360–63. doi:10.1002/anie.200703053. PMID 17847139.
- ^ Weeks 1968, p. 4.
- ^ a b Weeks 1968, p. 29.
- ^ a b c Weeks 1968, p. 31.
- ^ Bjorkman, Judith Kingston (1973). «Meteors and Meteorites in the ancient Near East». Meteoritics. 8 (2): 91–132. Bibcode:1973Metic…8…91B. doi:10.1111/j.1945-5100.1973.tb00146.x.
- ^ Comelli, Daniela; d’Orazio, Massimo; Folco, Luigi; El-Halwagy, Mahmud; Frizzi, Tommaso; Alberti, Roberto; Capogrosso, Valentina; Elnaggar, Abdelrazek; Hassan, Hala; Nevin, Austin; Porcelli, Franco; Rashed, Mohamed G; Valentini, Gianluca (2016). «The meteoritic origin of Tutankhamun’s iron dagger blade». Meteoritics & Planetary Science. 51 (7): 1301–09. Bibcode:2016M&PS…51.1301C. doi:10.1111/maps.12664.
- ^ Walsh, Declan (2 June 2016). «King Tut’s Dagger Made of ‘Iron From the Sky,’ Researchers Say». The New York Times. Archived from the original on 3 January 2022. Retrieved 4 June 2016.
the blade’s composition of iron, nickel and cobalt was an approximate match for a meteorite that landed in northern Egypt. The result «strongly suggests an extraterrestrial origin»
- ^ Ure, Andrew (1843). Technisches wörterbuch oder Handbuch der Gewerbskunde … : Bearb. nach Dr. Andrew Ure’s Dictionary of arts, manufactures and mines (in German). G. Haase. p. 492.
- ^ a b c d Weeks 1968, p. 32.
- ^ McNutt, Paula (1990 1). The Forging of Israel: Iron Technology, Symbolism and Tradition in Ancient Society. A&C Black.
- ^ Tewari, Rakesh. «The origins of Iron Working in India: New evidence from the Central Ganga plain and the Eastern Vindhyas» (PDF). State Archaeological Department. Retrieved 23 May 2010.
- ^ Photos, E. (1989). «The Question of Meteoritic versus Smelted Nickel-Rich Iron: Archaeological Evidence and Experimental Results». World Archaeology. Taylor & Francis, Ltd. 20 (3): 403–21. doi:10.1080/00438243.1989.9980081. JSTOR 124562.
- ^ Muhly, James D. (2003). «Metalworking/Mining in the Levant». In Lake, Richard Winona (ed.). Near Eastern Archaeology IN: Eisenbrauns. Vol. 180. pp. 174–83.
- ^ Witzel, Michael (2001), «Autochthonous Aryans? The Evidence from Old Indian and Iranian Texts», in Electronic Journal of Vedic Studies (EJVS) 7-3, pp. 1–93
- ^ Weeks, p. 33, quoting Cline, Walter (1937) «Mining and Metallurgy in Negro Africa,» George Banta Publishing Co., Menasha, Wis., pp. 17–23.
- ^ Riederer, Josef; Wartke, Ralf-B. (2009) «Iron», Cancik, Hubert; Schneider, Helmuth (eds.): Brill’s New Pauly, Brill.
- ^ Sawyer, Ralph D. and Sawyer, Mei-chün (1993). The Seven Military Classics of Ancient China. Boulder: Westview. ISBN 0-465-00304-4. p. 10.
- ^ Pigott, Vincent C. (1999). The Archaeometallurgy of the Asian Old World. Philadelphia: University of Pennsylvania Museum of Archaeology and Anthropology. ISBN 0-924171-34-0, p. 8.
- ^ Golas, Peter J. (1999). Science and Civilisation in China: Volume 5, Chemistry and Chemical Technology, Part 13, Mining. Cambridge University Press. p. 152. ISBN 978-0-521-58000-7.
earliest blast furnace discovered in China from about the first century AD
- ^ Pigott, Vincent C. (1999). The Archaeometallurgy of the Asian Old World. Philadelphia: University of Pennsylvania Museum of Archaeology and Anthropology. ISBN 0-924171-34-0, p. 191.
- ^ The Coming of the Ages of Steel. Brill Archive. 1961. p. 54.
- ^ Mott, R.A (2014). «Dry and Wet Puddling». Transactions of the Newcomen Society. 49: 156–57. doi:10.1179/tns.1977.011.
- ^ Wagner, Donald B. (2003). «Chinese blast furnaces from the 10th to the 14th century» (PDF). Historical Metallurgy. 37 (1): 25–37. Archived from the original (PDF) on 7 January 2018. Retrieved 7 January 2018. originally published in Wagner, Donald B. (2001). «Chinese blast furnaces from the 10th to the 14th century». West Asian Science, Technology, and Medicine. 18: 41–74. doi:10.1163/26669323-01801008.
- ^ Giannichedda, Enrico (2007): «Metal production in Late Antiquity», in Technology in Transition AD 300–650 Lavan, L.; Zanini, E. and Sarantis, A.(eds.), Brill, Leiden; ISBN 90-04-16549-5, p. 200.
- ^ a b c d e f g Biddle, Verne; Parker, Gregory. Chemistry, Precision and Design. A Beka Book, Inc.
- ^ Wagner, Donald B. (1993). Iron and Steel in Ancient China. Brill. pp. 335–340. ISBN 978-90-04-09632-5.
- ^ a b c d Greenwood & Earnshaw 1997, p. 1072.
- ^ Schivelbusch, G. (1986) The Railway Journey: Industrialization and Perception of Time and Space in the 19th Century. Oxford: Berg.
- ^ Spoerl, Joseph S. A Brief History of Iron and Steel Production Archived 2 June 2010 at the Wayback Machine. Saint Anselm College
- ^ Enghag, Per (8 January 2008). Encyclopedia of the Elements: Technical Data – History – Processing – Applications. pp. 190–91. ISBN 978-3-527-61234-5.
- ^ Whitaker, Robert D (1975). «An historical note on the conservation of mass». Journal of Chemical Education. 52 (10): 658. Bibcode:1975JChEd..52..658W. doi:10.1021/ed052p658.
- ^ Fontenrose, Joseph (1974). «Work, Justice, and Hesiod’s Five Ages». Classical Philology. 69 (1): 1–16. doi:10.1086/366027. JSTOR 268960. S2CID 161808359.
- ^ Schmidt, Eva (1981) Der preußische Eisenkunstguss. (Art of Prussian cast iron) Technik, Geschichte, Werke, Künstler. Verlag Mann, Berlin, ISBN 3-7861-1130-8
- ^ Lux, H. (1963) «Metallic Iron» in Handbook of Preparative Inorganic Chemistry, 2nd Ed. G. Brauer (ed.), Academic Press, NY. Vol. 2. pp. 1490–91.
- ^ Steel Statistical Yearbook 2010. World Steel Association
- ^ a b c d e f g Greenwood & Earnshaw 1997, p. 1073.
- ^ Song Yingxing (1637): The Tiangong Kaiwu encyclopedia.
- ^ Wang, Peng; Ryberg, Morten; Yang, Yi; Feng, Kuishuang; Kara, Sami; Hauschild, Michael; Chen, Wei-Qiang (6 April 2021). «Efficiency stagnation in global steel production urges joint supply- and demand-side mitigation efforts». Nature Communications. 12 (1): 2066. Bibcode:2021NatCo..12.2066W. doi:10.1038/s41467-021-22245-6. ISSN 2041-1723. PMC 8024266. PMID 33824307.
- ^ Verhoeven, J.D. (1975) Fundamentals of Physical Metallurgy, Wiley, New York, p. 326
- ^ Greenwood & Earnshaw 1997, pp. 1070–71.
- ^ a b Kohl, Walter H. (1995). Handbook of materials and techniques for vacuum devices. Springer. pp. 164–67. ISBN 1-56396-387-6.
- ^ a b Kuhn, Howard; Medlin, Dana; et al., eds. (2000). ASM Handbook – Mechanical Testing and Evaluation (PDF). Vol. 8. ASM International. p. 275. ISBN 0-87170-389-0. Archived from the original (PDF) on 9 February 2019. Retrieved 22 February 2022.
- ^ «Hardness Conversion Chart». Maryland Metrics. Archived from the original on 18 June 2015. Retrieved 23 May 2010.
- ^ Takaji, Kusakawa; Toshikatsu, Otani (1964). «Properties of Various Pure Irons: Study on pure iron I». Tetsu-to-Hagane. 50 (1): 42–47. doi:10.2355/tetsutohagane1955.50.1_42.
- ^ Raghavan, V. (2004). Materials Science and Engineering. PHI Learning Pvt. Ltd. p. 218. ISBN 81-203-2455-2.
- ^ Martin, John Wilson (2007). Concise encyclopedia of the structure of materials. Elsevier. p. 183. ISBN 978-0-08-045127-5.
- ^ a b Camp, James McIntyre; Francis, Charles Blaine (1920). The Making, Shaping and Treating of Steel. Pittsburgh: Carnegie Steel Company. pp. 173–74. ISBN 1-147-64423-3.
- ^ a b Smith, William F.; Hashemi, Javad (2006), Foundations of Materials Science and Engineering (4th ed.), McGraw-Hill, p. 431, ISBN 0-07-295358-6.
- ^ a b «Classification of Carbon and Low-Alloy Steels». Archived from the original on 2 January 2011. Retrieved 5 January 2008.
- ^ HSLA Steel, 15 November 2002, archived from the original on 30 December 2009, retrieved 11 October 2008.
- ^ Oberg, E.; et al. (1996), «Machinery’s Handbook», New York: Industrial Press (25th ed.), Industrial Press Inc: 440–42, Bibcode:1984msh..book…..R
- ^ Rokni, Sayed H.; Cossairt, J. Donald; Liu, James C. (January 2008). «Radiation Shielding at High-Energy Electron and Proton Accelerators» (PDF). Retrieved 6 August 2016.
- ^ a b c Greenwood & Earnshaw 1997, p. 1076.
- ^ Fürstner, Alois (2016). «Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes to Make This Base Metal a Multitasking Champion». ACS Central Science. 2 (11): 778–789. doi:10.1021/acscentsci.6b00272. PMC 5140022. PMID 27981231.
- ^ Bullock, R. Morris; et al. (2020). «Using nature’s blueprint to expand catalysis with Earth-abundant metals». Science. 369 (6505): eabc3183. doi:10.1126/science.abc3183. PMC 7875315. PMID 32792370.
- ^ Kolasinski, Kurt W. (2002). «Where are Heterogenous Reactions Important». Surface science: foundations of catalysis and nanoscience. John Wiley and Sons. pp. 15–16. ISBN 978-0-471-49244-3.
- ^ McKetta, John J. (1989). «Nitrobenzene and Nitrotoluene». Encyclopedia of Chemical Processing and Design: Volume 31 – Natural Gas Liquids and Natural Gasoline to Offshore Process Piping: High Performance Alloys. CRC Press. pp. 166–67. ISBN 978-0-8247-2481-8.
- ^ a b c Wildermuth, Egon; Stark, Hans; Friedrich, Gabriele; Ebenhöch, Franz Ludwig; Kühborth, Brigitte; Silver, Jack; Rituper, Rafael (2000). «Iron Compounds». Ullmann’s Encyclopedia of Industrial Chemistry. doi:10.1002/14356007.a14_591. ISBN 3-527-30673-0.
- ^ Stroud, Robert (1933). Diseases of Canaries. Canary Publishers Company. p. 203. ISBN 978-1-4465-4656-7.
- ^ World Health Organization (2021). World Health Organization model list of essential medicines: 22nd list (2021). Geneva: World Health Organization. hdl:10665/345533. WHO/MHP/HPS/EML/2021.02.
- ^ Dlouhy, Adrienne C.; Outten, Caryn E. (2013). Banci, Lucia (ed.). Metallomics and the Cell. Metal Ions in Life Sciences. Vol. 12. Springer. pp. 241–78. doi:10.1007/978-94-007-5561-1_8. ISBN 978-94-007-5560-4. PMC 3924584. PMID 23595675. electronic-book ISBN 978-94-007-5561-1
- ^
Yee, Gereon M.; Tolman, William B. (2015). Peter M.H. Kroneck; Martha E. Sosa Torres (eds.). Sustaining Life on Planet Earth: Metalloenzymes Mastering Dioxygen and Other Chewy Gases. Metal Ions in Life Sciences. Vol. 15. Springer. pp. 131–204. doi:10.1007/978-3-319-12415-5_5. PMID 25707468. - ^ a b c d e f g h i j k l m n o p Greenwood & Earnshaw 1997, pp. 1098–104.
- ^ Lippard, S.J.; Berg, J.M. (1994). Principles of Bioinorganic Chemistry. Mill Valley: University Science Books. ISBN 0-935702-73-3.
- ^ Kikuchi, G.; Yoshida, T.; Noguchi, M. (2005). «Heme oxygenase and heme degradation». Biochemical and Biophysical Research Communications. 338 (1): 558–67. doi:10.1016/j.bbrc.2005.08.020. PMID 16115609.
- ^
Uebe, René; Schüler, Dirk; «The Formation of Iron Biominerals «, pp 159-184 in «Metals, Microbes and Minerals: The Biogeochemical Side of Life» (2021) pp xiv + 341. Walter de Gruyter, Berlin. Editors Kroneck, Peter M.H. and Sosa Torres, Martha. DOI 10.1515/9783110589771-006 - ^ Neilands, J.B. (1995). «Siderophores: structure and function of microbial iron transport compounds». The Journal of Biological Chemistry. 270 (45): 26723–26. doi:10.1074/jbc.270.45.26723. PMID 7592901.
- ^ Neilands, J.B. (1981). «Microbial Iron Compounds». Annual Review of Biochemistry. 50 (1): 715–31. doi:10.1146/annurev.bi.50.070181.003435. PMID 6455965.
- ^ Boukhalfa, Hakim; Crumbliss, Alvin L. (2002). «Chemical aspects of siderophore mediated iron transport». BioMetals. 15 (4): 325–39. doi:10.1023/A:1020218608266. PMID 12405526. S2CID 19697776.
- ^ Nanami, M.; Ookawara, T.; Otaki, Y.; Ito, K.; Moriguchi, R.; Miyagawa, K.; Hasuike, Y.; Izumi, M.; Eguchi, H.; Suzuki, K.; Nakanishi, T. (2005). «Tumor necrosis factor-α-induced iron sequestration and oxidative stress in human endothelial cells». Arteriosclerosis, Thrombosis, and Vascular Biology. 25 (12): 2495–501. doi:10.1161/01.ATV.0000190610.63878.20. PMID 16224057.
- ^ Rouault, Tracey A. (2003). «How Mammals Acquire and Distribute Iron Needed for Oxygen-Based Metabolism». PLOS Biology. 1 (3): e9. doi:10.1371/journal.pbio.0000079. PMC 300689. PMID 14691550.
- ^ Boon EM, Downs A, Marcey D. «Proposed Mechanism of Catalase». Catalase: H2O2: H2O2 Oxidoreductase: Catalase Structural Tutorial. Retrieved 11 February 2007.
- ^ Boyington JC, Gaffney BJ, Amzel LM (1993). «The three-dimensional structure of an arachidonic acid 15-lipoxygenase». Science. 260 (5113): 1482–86. Bibcode:1993Sci…260.1482B. doi:10.1126/science.8502991. PMID 8502991.
- ^ Gray, N.K.; Hentze, M.W. (August 1994). «Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs». EMBO J. 13 (16): 3882–91. doi:10.1002/j.1460-2075.1994.tb06699.x. PMC 395301. PMID 8070415.
- ^ Gregory B. Vásquez; Xinhua Ji; Clara Fronticelli; Gary L. Gilliland (1998). «Human Carboxyhemoglobin at 2.2 Å Resolution: Structure and Solvent Comparisons of R-State, R2-State and T-State Hemoglobins». Acta Crystallogr. D. 54 (3): 355–66. doi:10.1107/S0907444997012250. PMID 9761903.
- ^ Sanderson, K (2017). «Mussels’ iron grip inspires strong and stretchy polymer». Chemical & Engineering News. American Chemical Society. 95 (44): 8. doi:10.1021/cen-09544-notw3. Retrieved 2 November 2017.
- ^ Food Standards Agency – Eat well, be well – Iron deficiency Archived 8 August 2006 at the Wayback Machine. Eatwell.gov.uk (5 March 2012). Retrieved on 27 June 2012.
- ^ Hoppe, M.; Hulthén, L.; Hallberg, L. (2005). «The relative bioavailability in humans of elemental iron powders for use in food fortification». European Journal of Nutrition. 45 (1): 37–44. doi:10.1007/s00394-005-0560-0. PMID 15864409. S2CID 42983904.
- ^ Pineda, O.; Ashmead, H. D. (2001). «Effectiveness of treatment of iron-deficiency anemia in infants and young children with ferrous bis-glycinate chelate». Nutrition. 17 (5): 381–4. doi:10.1016/S0899-9007(01)00519-6. PMID 11377130.
- ^ Ashmead, H. DeWayne (1989). Conversations on Chelation and Mineral Nutrition. Keats Publishing. ISBN 0-87983-501-X.
- ^ Institute of Medicine (US) Panel on Micronutrients (2001). «Iron» (PDF). Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Iron. National Academy Press. pp. 290–393. ISBN 0-309-07279-4. PMID 25057538. Archived from the original (PDF) on 9 September 2017. Retrieved 9 March 2017.
- ^ «Overview on Dietary Reference Values for the EU population as derived by the EFSA Panel on Dietetic Products, Nutrition and Allergies» (PDF). European Food Safety Authority. 2017.
- ^ «Tolerable Upper Intake Levels For Vitamins And Minerals» (PDF). European Food Safety Authority. 2006.
- ^ «Iron Deficiency Anemia». MediResource. Retrieved 17 December 2008.
- ^ Milman, N. (1996). «Serum ferritin in Danes: studies of iron status from infancy to old age, during blood donation and pregnancy». International Journal of Hematology. 63 (2): 103–35. doi:10.1016/0925-5710(95)00426-2. PMID 8867722.
- ^ «Federal Register May 27, 2016 Food Labeling: Revision of the Nutrition and Supplement Facts Labels. FR page 33982» (PDF).
- ^ «Daily Value Reference of the Dietary Supplement Label Database (DSLD)». Dietary Supplement Label Database (DSLD). Archived from the original on 7 April 2020. Retrieved 16 May 2020.
- ^ Centers for Disease Control and Prevention (2002). «Iron deficiency – United States, 1999–2000». MMWR. 51 (40): 897–99. PMID 12418542.
- ^ Hider, Robert C.; Kong, Xiaole (2013). «Chapter 8. Iron: Effect of Overload and Deficiency». In Astrid Sigel, Helmut Sigel and Roland K.O. Sigel (ed.). Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences. Vol. 13. Springer. pp. 229–94. doi:10.1007/978-94-007-7500-8_8. PMID 24470094.
- ^ Dlouhy, Adrienne C.; Outten, Caryn E. (2013). «Chapter 8.4 Iron Uptake, Trafficking and Storage». In Banci, Lucia (ed.). Metallomics and the Cell. Metal Ions in Life Sciences. Vol. 12. Springer. pp. 241–78. doi:10.1007/978-94-007-5561-1_8. ISBN 978-94-007-5560-4. PMC 3924584. PMID 23595675. electronic-book ISBN 978-94-007-5561-1
- ^ CDC Centers for Disease Control and Prevention (3 April 1998). «Recommendations to Prevent and Control Iron Deficiency in the United States». Morbidity and Mortality Weekly Report. 47 (RR3): 1. Retrieved 12 August 2014.
- ^ Centers for Disease Control and Prevention. «Iron and Iron Deficiency». Retrieved 12 August 2014.
- ^ Ramzi S. Cotran; Vinay Kumar; Tucker Collins; Stanley Leonard Robbins (1999). Robbins pathologic basis of disease. Saunders. ISBN 978-0-7216-7335-6. Retrieved 27 June 2012.
- ^ Ganz T (August 2003). «Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation». Blood. 102 (3): 783–8. doi:10.1182/blood-2003-03-0672. PMID 12663437. S2CID 28909635.
- ^ Durupt, S.; Durieu, I.; Nové-Josserand, R.; Bencharif, L.; Rousset, H.; Vital Durand, D. (2000). «Hereditary hemochromatosis». Rev Méd Interne. 21 (11): 961–71. doi:10.1016/S0248-8663(00)00252-6. PMID 11109593.
- ^ a b Cheney, K.; Gumbiner, C.; Benson, B.; Tenenbein, M. (1995). «Survival after a severe iron poisoning treated with intermittent infusions of deferoxamine». J Toxicol Clin Toxicol. 33 (1): 61–66. doi:10.3109/15563659509020217. PMID 7837315.
- ^ a b «Toxicity, Iron». Medscape. Retrieved 23 May 2010.
- ^ Dietary Reference Intakes (DRIs): Recommended Intakes for Individuals (PDF), Food and Nutrition Board, Institute of Medicine, National Academies, 2004, archived from the original (PDF) on 14 March 2013, retrieved 9 June 2009
- ^ Tenenbein, M. (1996). «Benefits of parenteral deferoxamine for acute iron poisoning». J Toxicol Clin Toxicol. 34 (5): 485–89. doi:10.3109/15563659609028005. PMID 8800185.
- ^ Wu H, Wu T, Xu X, Wang J, Wang J (May 2011). «Iron toxicity in mice with collagenase-induced intracerebral hemorrhage». J Cereb Blood Flow Metab. 31 (5): 1243–50. doi:10.1038/jcbfm.2010.209. PMC 3099628. PMID 21102602.
- ^ Robberecht, Harry; et al. (2020). «Magnesium, Iron, Zinc, Copper and Selenium Status in Attention-Deficit/Hyperactivity Disorder (ADHD)». Molecules. 25 (19): 4440. doi:10.3390/molecules25194440. PMC 7583976. PMID 32992575.
- ^ Soto-Insuga, V; et al. (2013). «[Role of iron in the treatment of attention deficit-hyperactivity disorder]». An Pediatr (Barc). 79 (4): 230–235. doi:10.1016/j.anpedi.2013.02.008. PMID 23582950.
- ^ Parisi, Pasquale; Villa, Maria Pia; Donfrancesco, Renato; Miano, Silvia; Paolino, Maria Chiara; Cortese, Samuele (August 2012). «Could treatment of iron deficiency both improve ADHD and reduce cardiovascular risk during treatment with ADHD drugs?». Medical Hypotheses. 79 (2): 246–249. doi:10.1016/j.mehy.2012.04.049. PMID 22632845.
- ^ Donfrancesco, Renato; Parisi, Pasquale; Vanacore, Nicola; Martines, Francesca; Sargentini, Vittorio; Cortese, Samuele (May 2013). «Iron and ADHD: Time to Move Beyond Serum Ferritin Levels». Journal of Attention Disorders. 17 (4): 347–357. doi:10.1177/1087054711430712. ISSN 1087-0547. PMID 22290693. S2CID 22445593.
- ^ Thévenod, Frank (2018). «Chapter 15. Iron and Its Role in Cancer Defense: A Double-Edged Sword». In Sigel, Astrid; Sigel, Helmut; Freisinger, Eva; Sigel, Roland K. O. (eds.). Metallo-Drugs: Development and Action of Anticancer Agents. Metal Ions in Life Sciences. Vol. 18. Berlin: de Gruyter GmbH. pp. 437–67. doi:10.1515/9783110470734-021. PMID 29394034.
- ^ a b Beguin, Y; Aapro, M; Ludwig, H; Mizzen, L; Osterborg, A (2014). «Epidemiological and nonclinical studies investigating effects of iron in carcinogenesis—a critical review». Critical Reviews in Oncology/Hematology. 89 (1): 1–15. doi:10.1016/j.critrevonc.2013.10.008. PMID 24275533.
- ^ Morel, F.M.M., Hudson, R.J.M., & Price, N.M. (1991). Limitation of productivity by trace metals in the sea. Limnology and Oceanography, 36(8), 1742-1755. doi:10.4319/lo.1991.36.8.1742
- ^ Brezezinski, M.A., Baines, S.B., Balch, W.M., Beucher, C.P., Chai, F., Dugdale, R.C., Krause, J.W., Landry, M.R., Marchi, A., Measures, C.I., Nelson, D.M., Parker, A.E., Poulton, A.J., Selph, K.E., Strutton, P.G., Taylor, A.G., & Twining, B.S.(2011). Co-limitation of diatoms by iron and silicic acid in the equatorial Pacific. Deep-Sea Research Part II: Topical Studies in Oceanography, 58(3-4), 493-511. doi:10.1016/j.dsr2.2010.08.005
- ^ Field, E. K., Kato, S., Findlay, A. J., MacDonald, D. J., Chiu, B. K., Luther, G. W., & Chan, C. S. (2016). Planktonic marine iron oxidizers drive iron mineralization under low-oxygen conditions. Geobiology, 14(5), 499-508. doi:10.1111/gbi.12189
- ^ Wells, M.L., Price, N.M., & Bruland, K.W. (1995). Iron chemistry in seawater and its relationship to phytoplankton: a workshop report. Marine Chemistry, 48(2), 157-182. doi:10.1016/0304-4203(94)00055-I
- ^ Lannuzel, D., Vancoppenolle, M., van der Merwe, P., de Jong, J., Meiners, K.M., Grotti, M., Nishioska, J., & Schoemann. (2016). Iron in sea ice: Review and new insights. Elementa: Science of the Anthropocene, 4 000130.
doi:10.12952/journal.elementa.000130 - ^ Raiswell, R. 2011. Iron Transport from the Continents to the Open Ocean: The Aging–Rejuvenation Cycle. Elements, 7(2), 101–106. doi:10.2113/gselements.7.2.101
- ^ Tagliabue, A., Bopp, L., Aumont,O., & Arrigo, K.R. (2009). Influence of light and temperature on the marine iron cycle: From theoretical to global modeling. Global Biogeochemical Cycles, 23.
doi:10.1029/2008GB003214
Bibliography
- Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 978-0-08-037941-8.
- Weeks, Mary Elvira; Leichester, Henry M. (1968). «Elements known to the ancients». Discovery of the elements. Easton, PA: Journal of Chemical Education. pp. 29–40. ISBN 0-7661-3872-0. LCCN 68-15217.
Further reading
- H.R. Schubert, History of the British Iron and Steel Industry … to 1775 AD (Routledge, London, 1957)
- R.F. Tylecote, History of Metallurgy (Institute of Materials, London 1992).
- R.F. Tylecote, «Iron in the Industrial Revolution» in J. Day and R.F. Tylecote, The Industrial Revolution in Metals (Institute of Materials 1991), 200–60.
External links
![]()
Wikiquote has quotations related to Iron.
![]()
Look up iron in Wiktionary, the free dictionary.
![]()
Wikimedia Commons has media related to Iron.
![]()
- It’s Elemental – Iron
- Iron at The Periodic Table of Videos (University of Nottingham)
- Metallurgy for the non-Metallurgist
- Iron by J.B. Calvert
This article is about the metallic element. For other uses, see Iron (disambiguation).
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Iron | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allotropes | see Allotropes of iron | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Appearance | lustrous metallic with a grayish tinge | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Standard atomic weight Ar°(Fe) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Iron in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 26 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Group | group 8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Period | period 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Block | d-block | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electron configuration | [Ar] 3d6 4s2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 8, 14, 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Physical properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phase at STP | solid | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Melting point | 1811 K (1538 °C, 2800 °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Boiling point | 3134 K (2862 °C, 5182 °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density (near r.t.) | 7.874 g/cm3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| when liquid (at m.p.) | 6.98 g/cm3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of fusion | 13.81 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of vaporization | 340 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar heat capacity | 25.10 J/(mol·K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Vapor pressure
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxidation states | −4, −2, −1, 0, +1,[2] +2, +3, +4, +5,[3] +6, +7[4] (an amphoteric oxide) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 1.83 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ionization energies |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic radius | empirical: 126 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Covalent radius | Low spin: 132±3 pm High spin: 152±6 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Van der Waals radius | 194 [1] pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Spectral lines of iron | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Natural occurrence | primordial | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure | body-centered cubic (bcc)
a=286.65 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure | face-centered cubic (fcc)
between 1185–1667 K; a=364.680 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Speed of sound thin rod | 5120 m/s (at r.t.) (electrolytic) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal expansion | 11.8 µm/(m⋅K) (at 25 °C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal conductivity | 80.4 W/(m⋅K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrical resistivity | 96.1 nΩ⋅m (at 20 °C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Curie point | 1043 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Magnetic ordering | ferromagnetic | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Young’s modulus | 211 GPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Shear modulus | 82 GPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bulk modulus | 170 GPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Poisson ratio | 0.29 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mohs hardness | 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Vickers hardness | 608 MPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Brinell hardness | 200–1180 MPa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAS Number | 7439-89-6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| History | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Discovery | before 5000 BC | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symbol | «Fe»: from Latin ferrum | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Main isotopes of iron
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| references |
Iron ( or ) is a chemical element with symbol Fe (from Latin: ferrum) and atomic number 26. It is a metal that belongs to the first transition series and group 8 of the periodic table. It is, by mass, the most common element on Earth, just ahead of oxygen (32.1% and 30.1%, respectively), forming much of Earth’s outer and inner core. It is the fourth most common element in the Earth’s crust, being mainly deposited by meteorites in its metallic state, with its ores also being found there.
Extracting usable metal from iron ores requires kilns or furnaces capable of reaching 1,500 °C (2,730 °F) or higher, about 500 °C (932 °F) higher than that required to smelt copper. Humans started to master that process in Eurasia during the 2nd millennium BCE and the use of iron tools and weapons began to displace copper alloys—in some regions, only around 1200 BCE. That event is considered the transition from the Bronze Age to the Iron Age. In the modern world, iron alloys, such as steel, stainless steel, cast iron and special steels, are by far the most common industrial metals, due to their mechanical properties and low cost. The iron and steel industry is thus very important economically, and iron is the cheapest metal, with a price of a few dollars per kilogram or pound.
Pristine and smooth pure iron surfaces are a mirror-like silvery-gray. Iron reacts readily with oxygen and water to produce brown-to-black hydrated iron oxides, commonly known as rust. Unlike the oxides of some other metals that form passivating layers, rust occupies more volume than the metal and thus flakes off, exposing more fresh surfaces for corrosion. High-purity irons (e.g. electrolytic iron) are more resistant to corrosion.
The body of an adult human contains about 4 grams (0.005% body weight) of iron, mostly in hemoglobin and myoglobin. These two proteins play essential roles in vertebrate metabolism, respectively oxygen transport by blood and oxygen storage in muscles. To maintain the necessary levels, human iron metabolism requires a minimum of iron in the diet. Iron is also the metal at the active site of many important redox enzymes dealing with cellular respiration and oxidation and reduction in plants and animals.[5]
Chemically, the most common oxidation states of iron are iron(II) and iron(III). Iron shares many properties of other transition metals, including the other group 8 elements, ruthenium and osmium. Iron forms compounds in a wide range of oxidation states, −2 to +7. Iron also forms many coordination compounds; some of them, such as ferrocene, ferrioxalate, and Prussian blue have substantial industrial, medical, or research applications.
Characteristics
Allotropes
Molar volume vs. pressure for α iron at room temperature
At least four allotropes of iron (differing atom arrangements in the solid) are known, conventionally denoted α, γ, δ, and ε.
The first three forms are observed at ordinary pressures. As molten iron cools past its freezing point of 1538 °C, it crystallizes into its δ allotrope, which has a body-centered cubic (bcc) crystal structure. As it cools further to 1394 °C, it changes to its γ-iron allotrope, a face-centered cubic (fcc) crystal structure, or austenite. At 912 °C and below, the crystal structure again becomes the bcc α-iron allotrope.[6]
The physical properties of iron at very high pressures and temperatures have also been studied extensively,[7][8] because of their relevance to theories about the cores of the Earth and other planets. Above approximately 10 GPa and temperatures of a few hundred kelvin or less, α-iron changes into another hexagonal close-packed (hcp) structure, which is also known as ε-iron. The higher-temperature γ-phase also changes into ε-iron, but does so at higher pressure.
Some controversial experimental evidence exists for a stable β phase at pressures above 50 GPa and temperatures of at least 1500 K. It is supposed to have an orthorhombic or a double hcp structure.[9] (Confusingly, the term «β-iron» is sometimes also used to refer to α-iron above its Curie point, when it changes from being ferromagnetic to paramagnetic, even though its crystal structure has not changed.[6])
The inner core of the Earth is generally presumed to consist of an iron-nickel alloy with ε (or β) structure.[10]
Melting and boiling points
The melting and boiling points of iron, along with its enthalpy of atomization, are lower than those of the earlier 3d elements from scandium to chromium, showing the lessened contribution of the 3d electrons to metallic bonding as they are attracted more and more into the inert core by the nucleus;[11] however, they are higher than the values for the previous element manganese because that element has a half-filled 3d sub-shell and consequently its d-electrons are not easily delocalized. This same trend appears for ruthenium but not osmium.[12]
The melting point of iron is experimentally well defined for pressures less than 50 GPa. For greater pressures, published data (as of 2007) still varies by tens of gigapascals and over a thousand kelvin.[13]
Magnetic properties
Magnetization curves of 9 ferromagnetic materials, showing saturation. 1. Sheet steel, 2. Silicon steel, 3. Cast steel, 4. Tungsten steel, 5. Magnet steel, 6. Cast iron, 7. Nickel, 8. Cobalt, 9. Magnetite[14]
Below its Curie point of 770 °C (1,420 °F; 1,040 K), α-iron changes from paramagnetic to ferromagnetic: the spins of the two unpaired electrons in each atom generally align with the spins of its neighbors, creating an overall magnetic field.[15] This happens because the orbitals of those two electrons (dz2 and dx2 −. y2) do not point toward neighboring atoms in the lattice, and therefore are not involved in metallic bonding.[6]
In the absence of an external source of magnetic field, the atoms get spontaneously partitioned into magnetic domains, about 10 micrometers across,[16] such that the atoms in each domain have parallel spins, but some domains have other orientations. Thus a macroscopic piece of iron will have a nearly zero overall magnetic field.
Application of an external magnetic field causes the domains that are magnetized in the same general direction to grow at the expense of adjacent ones that point in other directions, reinforcing the external field. This effect is exploited in devices that need to channel magnetic fields to fulfill design function, such as electrical transformers, magnetic recording heads, and electric motors. Impurities, lattice defects, or grain and particle boundaries can «pin» the domains in the new positions, so that the effect persists even after the external field is removed – thus turning the iron object into a (permanent) magnet.[15]
Similar behavior is exhibited by some iron compounds, such as the ferrites including the mineral magnetite, a crystalline form of the mixed iron(II,III) oxide Fe3O4 (although the atomic-scale mechanism, ferrimagnetism, is somewhat different). Pieces of magnetite with natural permanent magnetization (lodestones) provided the earliest compasses for navigation. Particles of magnetite were extensively used in magnetic recording media such as core memories, magnetic tapes, floppies, and disks, until they were replaced by cobalt-based materials.
Isotopes
Iron has four stable isotopes: 54Fe (5.845% of natural iron), 56Fe (91.754%), 57Fe (2.119%) and 58Fe (0.282%). 24 artificial isotopes have also been created. Of these stable isotopes, only 57Fe has a nuclear spin (−1⁄2). The nuclide 54Fe theoretically can undergo double electron capture to 54Cr, but the process has never been observed and only a lower limit on the half-life of 3.1×1022 years has been established.[17]
60Fe is an extinct radionuclide of long half-life (2.6 million years).[18] It is not found on Earth, but its ultimate decay product is its granddaughter, the stable nuclide 60Ni.[17] Much of the past work on isotopic composition of iron has focused on the nucleosynthesis of 60Fe through studies of meteorites and ore formation. In the last decade, advances in mass spectrometry have allowed the detection and quantification of minute, naturally occurring variations in the ratios of the stable isotopes of iron. Much of this work is driven by the Earth and planetary science communities, although applications to biological and industrial systems are emerging.[19]
In phases of the meteorites Semarkona and Chervony Kut, a correlation between the concentration of 60Ni, the granddaughter of 60Fe, and the abundance of the stable iron isotopes provided evidence for the existence of 60Fe at the time of formation of the Solar System. Possibly the energy released by the decay of 60Fe, along with that released by 26Al, contributed to the remelting and differentiation of asteroids after their formation 4.6 billion years ago. The abundance of 60Ni present in extraterrestrial material may bring further insight into the origin and early history of the Solar System.[20]
The most abundant iron isotope 56Fe is of particular interest to nuclear scientists because it represents the most common endpoint of nucleosynthesis.[21] Since 56Ni (14 alpha particles) is easily produced from lighter nuclei in the alpha process in nuclear reactions in supernovae (see silicon burning process), it is the endpoint of fusion chains inside extremely massive stars, since addition of another alpha particle, resulting in 60Zn, requires a great deal more energy. This 56Ni, which has a half-life of about 6 days, is created in quantity in these stars, but soon decays by two successive positron emissions within supernova decay products in the supernova remnant gas cloud, first to radioactive 56Co, and then to stable 56Fe. As such, iron is the most abundant element in the core of red giants, and is the most abundant metal in iron meteorites and in the dense metal cores of planets such as Earth.[22] It is also very common in the universe, relative to other stable metals of approximately the same atomic weight.[22][23] Iron is the sixth most abundant element in the universe, and the most common refractory element.[24]
Although a further tiny energy gain could be extracted by synthesizing 62Ni, which has a marginally higher binding energy than 56Fe, conditions in stars are unsuitable for this process. Element production in supernovas greatly favor iron over nickel, and in any case, 56Fe still has a lower mass per nucleon than 62Ni due to its higher fraction of lighter protons.[25] Hence, elements heavier than iron require a supernova for their formation, involving rapid neutron capture by starting 56Fe nuclei.[22]
In the far future of the universe, assuming that proton decay does not occur, cold fusion occurring via quantum tunnelling would cause the light nuclei in ordinary matter to fuse into 56Fe nuclei. Fission and alpha-particle emission would then make heavy nuclei decay into iron, converting all stellar-mass objects to cold spheres of pure iron.[26]
Origin and occurrence in nature
Cosmogenesis
Iron’s abundance in rocky planets like Earth is due to its abundant production during the runaway fusion and explosion of type Ia supernovae, which scatters the iron into space.[27][28]
Metallic iron
A polished and chemically etched piece of an iron meteorite, believed to be similar in composition to the Earth’s metallic core, showing individual crystals of the iron-nickel alloy (Widmanstatten pattern)
Metallic or native iron is rarely found on the surface of the Earth because it tends to oxidize. However, both the Earth’s inner and outer core, that account for 35% of the mass of the whole Earth, are believed to consist largely of an iron alloy, possibly with nickel. Electric currents in the liquid outer core are believed to be the origin of the Earth’s magnetic field. The other terrestrial planets (Mercury, Venus, and Mars) as well as the Moon are believed to have a metallic core consisting mostly of iron. The M-type asteroids are also believed to be partly or mostly made of metallic iron alloy.
The rare iron meteorites are the main form of natural metallic iron on the Earth’s surface. Items made of cold-worked meteoritic iron have been found in various archaeological sites dating from a time when iron smelting had not yet been developed; and the Inuit in Greenland have been reported to use iron from the Cape York meteorite for tools and hunting weapons.[29] About 1 in 20 meteorites consist of the unique iron-nickel minerals taenite (35–80% iron) and kamacite (90–95% iron).[30] Native iron is also rarely found in basalts that have formed from magmas that have come into contact with carbon-rich sedimentary rocks, which have reduced the oxygen fugacity sufficiently for iron to crystallize. This is known as Telluric iron and is described from a few localities, such as Disko Island in West Greenland, Yakutia in Russia and Bühl in Germany.[31]
Mantle minerals
Ferropericlase (Mg,Fe)O, a solid solution of periclase (MgO) and wüstite (FeO), makes up about 20% of the volume of the lower mantle of the Earth, which makes it the second most abundant mineral phase in that region after silicate perovskite (Mg,Fe)SiO3; it also is the major host for iron in the lower mantle.[32] At the bottom of the transition zone of the mantle, the reaction γ-(Mg,Fe)2[SiO4] ↔ (Mg,Fe)[SiO3] + (Mg,Fe)O transforms γ-olivine into a mixture of silicate perovskite and ferropericlase and vice versa. In the literature, this mineral phase of the lower mantle is also often called magnesiowüstite.[33] Silicate perovskite may form up to 93% of the lower mantle,[34] and the magnesium iron form, (Mg,Fe)SiO3, is considered to be the most abundant mineral in the Earth, making up 38% of its volume.[35]
Earth’s crust
While iron is the most abundant element on Earth, most of this iron is concentrated in the inner and outer cores.[36][37] The fraction of iron that is in Earth’s crust only amounts to about 5% of the overall mass of the crust and is thus only the fourth most abundant element in that layer (after oxygen, silicon, and aluminium).[38]
Most of the iron in the crust is combined with various other elements to form many iron minerals. An important class is the iron oxide minerals such as hematite (Fe2O3), magnetite (Fe3O4), and siderite (FeCO3), which are the major ores of iron. Many igneous rocks also contain the sulfide minerals pyrrhotite and pentlandite.[39][40] During weathering, iron tends to leach from sulfide deposits as the sulfate and from silicate deposits as the bicarbonate. Both of these are oxidized in aqueous solution and precipitate in even mildly elevated pH as iron(III) oxide.[41]
Banded iron formation in McKinley Park, Minnesota.
Large deposits of iron are banded iron formations, a type of rock consisting of repeated thin layers of iron oxides alternating with bands of iron-poor shale and chert. The banded iron formations were laid down in the time between 3,700 million years ago and 1,800 million years ago.[42][43]
Materials containing finely ground iron(III) oxides or oxide-hydroxides, such as ochre, have been used as yellow, red, and brown pigments since pre-historical times. They contribute as well to the color of various rocks and clays, including entire geological formations like the Painted Hills in Oregon and the Buntsandstein («colored sandstone», British Bunter).[44] Through Eisensandstein (a jurassic ‘iron sandstone’, e.g. from Donzdorf in Germany)[45] and Bath stone in the UK, iron compounds are responsible for the yellowish color of many historical buildings and sculptures.[46] The proverbial red color of the surface of Mars is derived from an iron oxide-rich regolith.[47]
Significant amounts of iron occur in the iron sulfide mineral pyrite (FeS2), but it is difficult to extract iron from it and it is therefore not exploited.[48] In fact, iron is so common that production generally focuses only on ores with very high quantities of it.[49]
According to the International Resource Panel’s Metal Stocks in Society report, the global stock of iron in use in society is 2,200 kg per capita. More-developed countries differ in this respect from less-developed countries (7,000–14,000 vs 2,000 kg per capita).[50]
Oceans
Ocean science demonstrated the role of the iron in the ancient seas in both marine biota and climate.[51]
Chemistry and compounds
| Oxidation state | Representative compound |
|---|---|
| −2 (d10) | Disodium tetracarbonylferrate (Collman’s reagent) |
| −1 (d9) | Fe 2(CO)2− 8 |
| 0 (d8) | Iron pentacarbonyl |
| 1 (d7) | Cyclopentadienyliron dicarbonyl dimer («Fp2«) |
| 2 (d6) | Ferrous sulfate, ferrocene |
| 3 (d5) | Ferric chloride, ferrocenium tetrafluoroborate |
| 4 (d4) | Fe(diars) 2Cl2+ 2, Ferryl tetrafluoroborate |
| 5 (d3) | FeO3− 4 |
| 6 (d2) | Potassium ferrate |
| 7 (d1) | [FeO4]– (matrix isolation, 4K) |
Iron shows the characteristic chemical properties of the transition metals, namely the ability to form variable oxidation states differing by steps of one and a very large coordination and organometallic chemistry: indeed, it was the discovery of an iron compound, ferrocene, that revolutionalized the latter field in the 1950s.[52] Iron is sometimes considered as a prototype for the entire block of transition metals, due to its abundance and the immense role it has played in the technological progress of humanity.[53] Its 26 electrons are arranged in the configuration [Ar]3d64s2, of which the 3d and 4s electrons are relatively close in energy, and thus a number of electrons can be ionized.[12]
Iron forms compounds mainly in the oxidation states +2 (iron(II), «ferrous») and +3 (iron(III), «ferric»). Iron also occurs in higher oxidation states, e.g., the purple potassium ferrate (K2FeO4), which contains iron in its +6 oxidation state. The anion [FeO4]– with iron in its +7 oxidation state, along with an iron(V)-peroxo isomer, has been detected by infrared spectroscopy at 4 K after cocondensation of laser-ablated Fe atoms with a mixture of O2/Ar.[54] Iron(IV) is a common intermediate in many biochemical oxidation reactions.[55][56] Numerous organoiron compounds contain formal oxidation states of +1, 0, −1, or even −2. The oxidation states and other bonding properties are often assessed using the technique of Mössbauer spectroscopy.[57] Many mixed valence compounds contain both iron(II) and iron(III) centers, such as magnetite and Prussian blue (Fe4(Fe[CN]6)3).[56] The latter is used as the traditional «blue» in blueprints.[58]
Iron is the first of the transition metals that cannot reach its group oxidation state of +8, although its heavier congeners ruthenium and osmium can, with ruthenium having more difficulty than osmium.[6] Ruthenium exhibits an aqueous cationic chemistry in its low oxidation states similar to that of iron, but osmium does not, favoring high oxidation states in which it forms anionic complexes.[6] In the second half of the 3d transition series, vertical similarities down the groups compete with the horizontal similarities of iron with its neighbors cobalt and nickel in the periodic table, which are also ferromagnetic at room temperature and share similar chemistry. As such, iron, cobalt, and nickel are sometimes grouped together as the iron triad.[53]
Unlike many other metals, iron does not form amalgams with mercury. As a result, mercury is traded in standardized 76 pound flasks (34 kg) made of iron.[59]
Iron is by far the most reactive element in its group; it is pyrophoric when finely divided and dissolves easily in dilute acids, giving Fe2+. However, it does not react with concentrated nitric acid and other oxidizing acids due to the formation of an impervious oxide layer, which can nevertheless react with hydrochloric acid.[6] High-purity iron, called electrolytic iron, is considered to be resistant to rust, due to its oxide layer.
Binary compounds
Oxides and sulfides
Ferrous or iron(II) oxide,
FeO
Ferric or iron(III) oxide
Fe2O3
Ferrosoferric or iron(II,III) oxide
Fe3O4
Iron forms various oxide and hydroxide compounds; the most common are iron(II,III) oxide (Fe3O4), and iron(III) oxide (Fe2O3). Iron(II) oxide also exists, though it is unstable at room temperature. Despite their names, they are actually all non-stoichiometric compounds whose compositions may vary.[60] These oxides are the principal ores for the production of iron (see bloomery and blast furnace). They are also used in the production of ferrites, useful magnetic storage media in computers, and pigments. The best known sulfide is iron pyrite (FeS2), also known as fool’s gold owing to its golden luster.[56] It is not an iron(IV) compound, but is actually an iron(II) polysulfide containing Fe2+ and S2−
2 ions in a distorted sodium chloride structure.[60]
Halides
Hydrated iron(III) chloride (ferric chloride)
The binary ferrous and ferric halides are well-known. The ferrous halides typically arise from treating iron metal with the corresponding hydrohalic acid to give the corresponding hydrated salts.[56]
- Fe + 2 HX → FeX2 + H2 (X = F, Cl, Br, I)
Iron reacts with fluorine, chlorine, and bromine to give the corresponding ferric halides, ferric chloride being the most common.[61]
- 2 Fe + 3 X2 → 2 FeX3 (X = F, Cl, Br)
Ferric iodide is an exception, being thermodynamically unstable due to the oxidizing power of Fe3+ and the high reducing power of I−:[61]
- 2 I− + 2 Fe3+ → I2 + 2 Fe2+ (E0 = +0.23 V)
Ferric iodide, a black solid, is not stable in ordinary conditions, but can be prepared through the reaction of iron pentacarbonyl with iodine and carbon monoxide in the presence of hexane and light at the temperature of −20 °C, with oxygen and water excluded.[61]Complexes of ferric iodide with some soft bases are known to be stable compounds.[62][63]
Solution chemistry
Comparison of colors of solutions of ferrate (left) and permanganate (right)
The standard reduction potentials in acidic aqueous solution for some common iron ions are given below:[6]
| [Fe(H2O)6]2+ + 2 e− | ⇌ Fe | E0 = −0.447 V |
| [Fe(H2O)6]3+ + e− | ⇌ [Fe(H2O)6]2+ | E0 = +0.77 V |
| FeO2− 4 + 8 H3O+ + 3 e− | ⇌ [Fe(H2O)6]3+ + 6 H2O | E0 = +2.20 V |
The red-purple tetrahedral ferrate(VI) anion is such a strong oxidizing agent that it oxidizes ammonia to nitrogen (N2) and water to oxygen[61]
- 4 FeO2−
4 + 34 H
2O → 4 [Fe(H2O)6]3+ + 20 OH−
+ 3 O2
The pale-violet hexaquo complex [Fe(H2O)6]3+ is an acid such that above above pH 0 it is fully hydrolyzed:[64]
| [Fe(H2O)6]3+ | ⇌ [Fe(H2O)5(OH)]2+ + H+ | K = 10−3.05 mol dm−3 |
| [Fe(H2O)5(OH)]2+ | ⇌ [Fe(H2O)4(OH)2]+ + H+ | K = 10−3.26 mol dm−3 |
| 2[Fe(H2O)6]3+ | ⇌ [Fe(H2O)4(OH)]4+2 + 2H+ + 2H2O | K = 10−2.91 mol dm−3 |
As pH rises above 0 the above yellow hydrolyzed species form and as it rises above 2–3, reddish-brown hydrous iron(III) oxide precipitates out of solution. Although Fe3+ has a d5 configuration, its absorption spectrum is not like that of Mn2+ with its weak, spin-forbidden d–d bands, because Fe3+ has higher positive charge and is more polarizing, lowering the energy of its ligand-to-metal charge transfer absorptions. Thus, all the above complexes are rather strongly colored, with the single exception of the hexaquo ion – and even that has a spectrum dominated by charge transfer in the near ultraviolet region.[64] On the other hand, the pale green iron(II) hexaquo ion [Fe(H2O)6]2+ does not undergo appreciable hydrolysis. Carbon dioxide is not evolved when carbonate anions are added, which instead results in white iron(II) carbonate being precipitated out. In excess carbon dioxide this forms the slightly soluble bicarbonate, which occurs commonly in groundwater, but it oxidises quickly in air to form iron(III) oxide that accounts for the brown deposits present in a sizeable number of streams.[65]
Coordination compounds
Due to its electronic structure, iron has a very large coordination and organometallic chemistry.
Many coordination compounds of iron are known. A typical six-coordinate anion is hexachloroferrate(III), [FeCl6]3−, found in the mixed salt tetrakis(methylammonium) hexachloroferrate(III) chloride.[66][67] Complexes with multiple bidentate ligands have geometric isomers. For example, the trans-chlorohydridobis(bis-1,2-(diphenylphosphino)ethane)iron(II) complex is used as a starting material for compounds with the Fe(dppe)2 moiety.[68][69] The ferrioxalate ion with three oxalate ligands (shown at right) displays helical chirality with its two non-superposable geometries labelled Λ (lambda) for the left-handed screw axis and Δ (delta) for the right-handed screw axis, in line with IUPAC conventions.[64] Potassium ferrioxalate is used in chemical actinometry and along with its sodium salt undergoes photoreduction applied in old-style photographic processes. The dihydrate of iron(II) oxalate has a polymeric structure with co-planar oxalate ions bridging between iron centres with the water of crystallisation located forming the caps of each octahedron, as illustrated below.[70]
Crystal structure of iron(II) oxalate dihydrate, showing iron (gray), oxygen (red), carbon (black), and hydrogen (white) atoms.
Blood-red positive thiocyanate test for iron(III)
Iron(III) complexes are quite similar to those of chromium(III) with the exception of iron(III)’s preference for O-donor instead of N-donor ligands. The latter tend to be rather more unstable than iron(II) complexes and often dissociate in water. Many Fe–O complexes show intense colors and are used as tests for phenols or enols. For example, in the ferric chloride test, used to determine the presence of phenols, iron(III) chloride reacts with a phenol to form a deep violet complex:[64]
- 3 ArOH + FeCl3 → Fe(OAr)3 + 3 HCl (Ar = aryl)
Among the halide and pseudohalide complexes, fluoro complexes of iron(III) are the most stable, with the colorless [FeF5(H2O)]2− being the most stable in aqueous solution. Chloro complexes are less stable and favor tetrahedral coordination as in [FeCl4]−; [FeBr4]− and [FeI4]− are reduced easily to iron(II). Thiocyanate is a common test for the presence of iron(III) as it forms the blood-red [Fe(SCN)(H2O)5]2+. Like manganese(II), most iron(III) complexes are high-spin, the exceptions being those with ligands that are high in the spectrochemical series such as cyanide. An example of a low-spin iron(III) complex is [Fe(CN)6]3−. Iron shows a great variety of electronic spin states, including every possible spin quantum number value for a d-block element from 0 (diamagnetic) to 5⁄2 (5 unpaired electrons). This value is always half the number of unpaired electrons. Complexes with zero to two unpaired electrons are considered low-spin and those with four or five are considered high-spin.[60]
Iron(II) complexes are less stable than iron(III) complexes but the preference for O-donor ligands is less marked, so that for example [Fe(NH3)6]2+ is known while [Fe(NH3)6]3+ is not. They have a tendency to be oxidized to iron(III) but this can be moderated by low pH and the specific ligands used.[65]
Organometallic compounds
Organoiron chemistry is the study of organometallic compounds of iron, where carbon atoms are covalently bound to the metal atom. They are many and varied, including cyanide complexes, carbonyl complexes, sandwich and half-sandwich compounds.
![]()
Prussian blue or «ferric ferrocyanide», Fe4[Fe(CN)6]3, is an old and well-known iron-cyanide complex, extensively used as pigment and in several other applications. Its formation can be used as a simple wet chemistry test to distinguish between aqueous solutions of Fe2+ and Fe3+ as they react (respectively) with potassium ferricyanide and potassium ferrocyanide to form Prussian blue.[56]
Another old example of an organoiron compound is iron pentacarbonyl, Fe(CO)5, in which a neutral iron atom is bound to the carbon atoms of five carbon monoxide molecules. The compound can be used to make carbonyl iron powder, a highly reactive form of metallic iron. Thermolysis of iron pentacarbonyl gives triiron dodecacarbonyl, Fe3(CO)12, a complex with a cluster of three iron atoms at its core. Collman’s reagent, disodium tetracarbonylferrate, is a useful reagent for organic chemistry; it contains iron in the −2 oxidation state. Cyclopentadienyliron dicarbonyl dimer contains iron in the rare +1 oxidation state.[71]
Structural formula of ferrocene and a powdered sample
A landmark in this field was the discovery in 1951 of the remarkably stable sandwich compound ferrocene Fe(C5H5)2, by Pauson and Kealy[72] and independently by Miller and colleagues,[73] whose surprising molecular structure was determined only a year later by Woodward and Wilkinson[74] and Fischer.[75]
Ferrocene is still one of the most important tools and models in this class.[76]
Iron-centered organometallic species are used as catalysts. The Knölker complex, for example, is a transfer hydrogenation catalyst for ketones.[77]
Industrial uses
The iron compounds produced on the largest scale in industry are iron(II) sulfate (FeSO4·7H2O) and iron(III) chloride (FeCl3). The former is one of the most readily available sources of iron(II), but is less stable to aerial oxidation than Mohr’s salt ((NH4)2Fe(SO4)2·6H2O). Iron(II) compounds tend to be oxidized to iron(III) compounds in the air.[56]
History
Development of iron metallurgy
Iron is one of the elements undoubtedly known to the ancient world.[78] It has been worked, or wrought, for millennia. However, iron artefacts of great age are much rarer than objects made of gold or silver due to the ease with which iron corrodes.[79] The technology developed slowly, and even after the discovery of smelting it took many centuries for iron to replace bronze as the metal of choice for tools and weapons.
Meteoritic iron
Beads made from meteoric iron in 3500 BC or earlier were found in Gerzeh, Egypt by G.A. Wainwright.[80] The beads contain 7.5% nickel, which is a signature of meteoric origin since iron found in the Earth’s crust generally has only minuscule nickel impurities.
Meteoric iron was highly regarded due to its origin in the heavens and was often used to forge weapons and tools.[80] For example, a dagger made of meteoric iron was found in the tomb of Tutankhamun, containing similar proportions of iron, cobalt, and nickel to a meteorite discovered in the area, deposited by an ancient meteor shower.[81][82][83] Items that were likely made of iron by Egyptians date from 3000 to 2500 BC.[79]
Meteoritic iron is comparably soft and ductile and easily cold forged but may get brittle when heated because of the nickel content.[84]
Wrought iron
The symbol for Mars has been used since antiquity to represent iron.
The iron pillar of Delhi is an example of the iron extraction and processing methodologies of early India.
The first iron production started in the Middle Bronze Age, but it took several centuries before iron displaced bronze. Samples of smelted iron from Asmar, Mesopotamia and Tall Chagar Bazaar in northern Syria were made sometime between 3000 and 2700 BC.[85] The Hittites established an empire in north-central Anatolia around 1600 BC. They appear to be the first to understand the production of iron from its ores and regard it highly in their society.[86] The Hittites began to smelt iron between 1500 and 1200 BC and the practice spread to the rest of the Near East after their empire fell in 1180 BC.[85] The subsequent period is called the Iron Age.
Artifacts of smelted iron are found in India dating from 1800 to 1200 BC,[87] and in the Levant from about 1500 BC (suggesting smelting in Anatolia or the Caucasus).[88][89] Alleged references (compare history of metallurgy in South Asia) to iron in the Indian Vedas have been used for claims of a very early usage of iron in India respectively to date the texts as such. The rigveda term ayas (metal) refers to copper, while iron which is called as śyāma ayas, literally «black copper», first is mentioned in the post-rigvedic Atharvaveda.[90]
Some archaeological evidence suggests iron was smelted in Zimbabwe and southeast Africa as early as the eighth century BC.[91] Iron working was introduced to Greece in the late 11th century BC, from which it spread quickly throughout Europe.[92]
Iron sickle from Ancient Greece.
The spread of ironworking in Central and Western Europe is associated with Celtic expansion. According to Pliny the Elder, iron use was common in the Roman era.[80] In the lands of what is now considered China, iron appears approximately 700–500 BC.[93] Iron smelting may have been introduced into China through Central Asia.[94] The earliest evidence of the use of a blast furnace in China dates to the 1st century AD,[95] and cupola furnaces were used as early as the Warring States period (403–221 BC).[96] Usage of the blast and cupola furnace remained widespread during the Tang and Song dynasties.[97]
During the Industrial Revolution in Britain, Henry Cort began refining iron from pig iron to wrought iron (or bar iron) using innovative production systems. In 1783 he patented the puddling process for refining iron ore. It was later improved by others, including Joseph Hall.[98]
Cast iron
Cast iron was first produced in China during 5th century BC,[99] but was hardly in Europe until the medieval period.[100][101] The earliest cast iron artifacts were discovered by archaeologists in what is now modern Luhe County, Jiangsu in China. Cast iron was used in ancient China for warfare, agriculture, and architecture.[102] During the medieval period, means were found in Europe of producing wrought iron from cast iron (in this context known as pig iron) using finery forges. For all these processes, charcoal was required as fuel.[103]
Medieval blast furnaces were about 10 feet (3.0 m) tall and made of fireproof brick; forced air was usually provided by hand-operated bellows.[101] Modern blast furnaces have grown much bigger, with hearths fourteen meters in diameter that allow them to produce thousands of tons of iron each day, but essentially operate in much the same way as they did during medieval times.[103]
In 1709, Abraham Darby I established a coke-fired blast furnace to produce cast iron, replacing charcoal, although continuing to use blast furnaces. The ensuing availability of inexpensive iron was one of the factors leading to the Industrial Revolution. Toward the end of the 18th century, cast iron began to replace wrought iron for certain purposes, because it was cheaper. Carbon content in iron was not implicated as the reason for the differences in properties of wrought iron, cast iron, and steel until the 18th century.[85]
Since iron was becoming cheaper and more plentiful, it also became a major structural material following the building of the innovative first iron bridge in 1778. This bridge still stands today as a monument to the role iron played in the Industrial Revolution. Following this, iron was used in rails, boats, ships, aqueducts, and buildings, as well as in iron cylinders in steam engines.[103] Railways have been central to the formation of modernity and ideas of progress[104] and various languages refer to railways as iron road (e.g. French chemin de fer, German Eisenbahn, Turkish demiryolu, Russian железная дорога, Chinese, Japanese, and Korean 鐵道, Vietnamese đường sắt).
Steel
Steel (with smaller carbon content than pig iron but more than wrought iron) was first produced in antiquity by using a bloomery. Blacksmiths in Luristan in western Persia were making good steel by 1000 BC.[85] Then improved versions, Wootz steel by India and Damascus steel were developed around 300 BC and AD 500 respectively. These methods were specialized, and so steel did not become a major commodity until the 1850s.[105]
New methods of producing it by carburizing bars of iron in the cementation process were devised in the 17th century. In the Industrial Revolution, new methods of producing bar iron without charcoal were devised and these were later applied to produce steel. In the late 1850s, Henry Bessemer invented a new steelmaking process, involving blowing air through molten pig iron, to produce mild steel. This made steel much more economical, thereby leading to wrought iron no longer being produced in large quantities.[106]
Foundations of modern chemistry
In 1774, Antoine Lavoisier used the reaction of water steam with metallic iron inside an incandescent iron tube to produce hydrogen in his experiments leading to the demonstration of the conservation of mass, which was instrumental in changing chemistry from a qualitative science to a quantitative one.[107]
Symbolic role
«Gold gab ich für Eisen» – «I gave gold for iron». German-American brooch from WWI.
Iron plays a certain role in mythology and has found various usage as a metaphor and in folklore. The Greek poet Hesiod’s Works and Days (lines 109–201) lists different ages of man named after metals like gold, silver, bronze and iron to account for successive ages of humanity.[108] The Iron Age was closely related with Rome, and in Ovid’s Metamorphoses
The Virtues, in despair, quit the earth; and the depravity of man becomes universal and complete. Hard steel succeeded then.
An example of the importance of iron’s symbolic role may be found in the German Campaign of 1813. Frederick William III commissioned then the first Iron Cross as military decoration. Berlin iron jewellery reached its peak production between 1813 and 1815, when the Prussian royal family urged citizens to donate gold and silver jewellery for military funding. The inscription Gold gab ich für Eisen (I gave gold for iron) was used as well in later war efforts.[109]
Production of metallic iron
Iron furnace in Columbus, Ohio, 1922
Laboratory routes
For a few limited purposes when it is needed, pure iron is produced in the laboratory in small quantities by reducing the pure oxide or hydroxide with hydrogen, or forming iron pentacarbonyl and heating it to 250 °C so that it decomposes to form pure iron powder.[41] Another method is electrolysis of ferrous chloride onto an iron cathode.[110]
Main industrial route
| Country | Iron ore | Pig iron | Direct iron | Steel |
|---|---|---|---|---|
| 1,114.9 | 549.4 | 573.6 | ||
| 393.9 | 4.4 | 5.2 | ||
| 305.0 | 25.1 | 0.011 | 26.5 | |
| 66.9 | 87.5 | |||
| 257.4 | 38.2 | 23.4 | 63.5 | |
| 92.1 | 43.9 | 4.7 | 60.0 | |
| 65.8 | 25.7 | 29.9 | ||
| 0.1 | 27.3 | 48.6 | ||
| 0.4 | 20.1 | 0.38 | 32.7 | |
| World | 1,594.9 | 914.0 | 64.5 | 1,232.4 |
Nowadays, the industrial production of iron or steel consists of two main stages. In the first stage, iron ore is reduced with coke in a blast furnace, and the molten metal is separated from gross impurities such as silicate minerals. This stage yields an alloy—pig iron—that contains relatively large amounts of carbon. In the second stage, the amount of carbon in the pig iron is lowered by oxidation to yield wrought iron, steel, or cast iron.[112] Other metals can be added at this stage to form alloy steels.
17th century Chinese illustration of workers at a blast furnace, making wrought iron from pig iron[113]
How iron was extracted in the 19th century
Blast furnace processing
The blast furnace is loaded with iron ores, usually hematite Fe2O3 or magnetite Fe3O4, along with coke (coal that has been separately baked to remove volatile components) and flux (limestone or dolomite). «Blasts» of air pre-heated to 900 °C (sometimes with oxygen enrichment) is blown through the mixture, in sufficient amount to turn the carbon into carbon monoxide:[112]
This reaction raises the temperature to about 2000 °C. The carbon monoxide reduces the iron ore to metallic iron[112]
Some iron in the high-temperature lower region of the furnace reacts directly with the coke:[112]
The flux removes silicaceous minerals in the ore, which would otherwise clog the furnace: The heat of the furnace decomposes the carbonates to calcium oxide, which reacts with any excess silica to form a slag composed of calcium silicate CaSiO3 or other products. At the furnace’s temperature, the metal and the slag are both molten. They collect at the bottom as two immiscible liquid layers (with the slag on top), that are then easily separated.[112] The slag can be used as a material in road construction or to improve mineral-poor soils for agriculture.[101]
Steelmaking thus remains one of the largest industrial contributors of CO2 emissions in the world.[114]
This heap of iron ore pellets will be used in steel production.
Steelmaking
A pot of molten iron being used to make steel
The pig iron produced by the blast furnace process contains up to 4–5% carbon (by mass), with small amounts of other impurities like sulfur, magnesium, phosphorus, and manganese. This high level of carbon makes it relatively weak and brittle. Reducing the amount of carbon to 0.002–2.1% produces steel, which may be up to 1000 times harder than pure iron. A great variety of steel articles can then be made by cold working, hot rolling, forging, machining, etc. Removing the impurities from pig iron, but leaving 2–4% carbon, results in cast iron, which is cast by foundries into articles such as stoves, pipes, radiators, lamp-posts, and rails.[112]
Steel products often undergo various heat treatments after they are forged to shape. Annealing consists of heating them to 700–800 °C for several hours and then gradual cooling. It makes the steel softer and more workable.[115]
Direct iron reduction
Owing to environmental concerns, alternative methods of processing iron have been developed. «Direct iron reduction» reduces iron ore to a ferrous lump called «sponge» iron or «direct» iron that is suitable for steelmaking.[101] Two main reactions comprise the direct reduction process:
Natural gas is partially oxidized (with heat and a catalyst):[101]
Iron ore is then treated with these gases in a furnace, producing solid sponge iron:[101]
Silica is removed by adding a limestone flux as described above.[101]
Thermite process
Ignition of a mixture of aluminium powder and iron oxide yields metallic iron via the thermite reaction:
Alternatively pig iron may be made into steel (with up to about 2% carbon) or wrought iron (commercially pure iron). Various processes have been used for this, including finery forges, puddling furnaces, Bessemer converters, open hearth furnaces, basic oxygen furnaces, and electric arc furnaces. In all cases, the objective is to oxidize some or all of the carbon, together with other impurities. On the other hand, other metals may be added to make alloy steels.[103]
Applications
As structural material
Iron is the most widely used of all the metals, accounting for over 90% of worldwide metal production. Its low cost and high strength often make it the material of choice to withstand stress or transmit forces, such as the construction of machinery and machine tools, rails, automobiles, ship hulls, concrete reinforcing bars, and the load-carrying framework of buildings. Since pure iron is quite soft, it is most commonly combined with alloying elements to make steel.[116]
Mechanical properties
| Material | TS (MPa) | BH (Brinell) |
|---|---|---|
| Iron whiskers | 11000 | |
| Ausformed (hardened) steel | 2930 | 850–1200 |
| Martensitic steel | 2070 | 600 |
| Bainitic steel | 1380 | 400 |
| Pearlitic steel | 1200 | 350 |
| Cold-worked iron | 690 | 200 |
| Small-grain iron | 340 | 100 |
| Carbon-containing iron | 140 | 40 |
| Pure, single-crystal iron | 10 | 3 |
The mechanical properties of iron and its alloys are extremely relevant to their structural applications. Those properties can be evaluated in various ways, including the Brinell test, the Rockwell test and the Vickers hardness test.
The properties of pure iron are often used to calibrate measurements or to compare tests.[118][119] However, the mechanical properties of iron are significantly affected by the sample’s purity: pure, single crystals of iron are actually softer than aluminium,[117] and the purest industrially produced iron (99.99%) has a hardness of 20–30 Brinell.[120] The pure iron (99.9%~99.999%), especially called electrolytic iron, is industrially produced by electrolytic refining.
An increase in the carbon content will cause a significant increase in the hardness and tensile strength of iron. Maximum hardness of 65 Rc is achieved with a 0.6% carbon content, although the alloy has low tensile strength.[121] Because of the softness of iron, it is much easier to work with than its heavier congeners ruthenium and osmium.[12]
Iron-carbon phase diagram
Types of steels and alloys
α-Iron is a fairly soft metal that can dissolve only a small concentration of carbon (no more than 0.021% by mass at 910 °C).[122] Austenite (γ-iron) is similarly soft and metallic but can dissolve considerably more carbon (as much as 2.04% by mass at 1146 °C). This form of iron is used in the type of stainless steel used for making cutlery, and hospital and food-service equipment.[16]
Commercially available iron is classified based on purity and the abundance of additives. Pig iron has 3.5–4.5% carbon[123] and contains varying amounts of contaminants such as sulfur, silicon and phosphorus. Pig iron is not a saleable product, but rather an intermediate step in the production of cast iron and steel. The reduction of contaminants in pig iron that negatively affect material properties, such as sulfur and phosphorus, yields cast iron containing 2–4% carbon, 1–6% silicon, and small amounts of manganese.[112] Pig iron has a melting point in the range of 1420–1470 K, which is lower than either of its two main components, and makes it the first product to be melted when carbon and iron are heated together.[6] Its mechanical properties vary greatly and depend on the form the carbon takes in the alloy.[12]
«White» cast irons contain their carbon in the form of cementite, or iron carbide (Fe3C).[12] This hard, brittle compound dominates the mechanical properties of white cast irons, rendering them hard, but unresistant to shock. The broken surface of a white cast iron is full of fine facets of the broken iron carbide, a very pale, silvery, shiny material, hence the appellation. Cooling a mixture of iron with 0.8% carbon slowly below 723 °C to room temperature results in separate, alternating layers of cementite and α-iron, which is soft and malleable and is called pearlite for its appearance. Rapid cooling, on the other hand, does not allow time for this separation and creates hard and brittle martensite. The steel can then be tempered by reheating to a temperature in between, changing the proportions of pearlite and martensite. The end product below 0.8% carbon content is a pearlite-αFe mixture, and that above 0.8% carbon content is a pearlite-cementite mixture.[12]
In gray iron the carbon exists as separate, fine flakes of graphite, and also renders the material brittle due to the sharp edged flakes of graphite that produce stress concentration sites within the material.[124] A newer variant of gray iron, referred to as ductile iron, is specially treated with trace amounts of magnesium to alter the shape of graphite to spheroids, or nodules, reducing the stress concentrations and vastly increasing the toughness and strength of the material.[124]
Wrought iron contains less than 0.25% carbon but large amounts of slag that give it a fibrous characteristic.[123] It is a tough, malleable product, but not as fusible as pig iron. If honed to an edge, it loses it quickly. Wrought iron is characterized by the presence of fine fibers of slag entrapped within the metal. Wrought iron is more corrosion resistant than steel. It has been almost completely replaced by mild steel for traditional «wrought iron» products and blacksmithing.
Mild steel corrodes more readily than wrought iron, but is cheaper and more widely available. Carbon steel contains 2.0% carbon or less,[125] with small amounts of manganese, sulfur, phosphorus, and silicon. Alloy steels contain varying amounts of carbon as well as other metals, such as chromium, vanadium, molybdenum, nickel, tungsten, etc. Their alloy content raises their cost, and so they are usually only employed for specialist uses. One common alloy steel, though, is stainless steel. Recent developments in ferrous metallurgy have produced a growing range of microalloyed steels, also termed ‘HSLA’ or high-strength, low alloy steels, containing tiny additions to produce high strengths and often spectacular toughness at minimal cost.[125][126][127]
Alloys with high purity elemental makeups (such as alloys of electrolytic iron) have specifically enhanced properties such as ductility, tensile strength, toughness, fatigue strength, heat resistance, and corrosion resistance.
Apart from traditional applications, iron is also used for protection from ionizing radiation. Although it is lighter than another traditional protection material, lead, it is much stronger mechanically. The attenuation of radiation as a function of energy is shown in the graph.[128]
The main disadvantage of iron and steel is that pure iron, and most of its alloys, suffer badly from rust if not protected in some way, a cost amounting to over 1% of the world’s economy.[129] Painting, galvanization, passivation, plastic coating and bluing are all used to protect iron from rust by excluding water and oxygen or by cathodic protection. The mechanism of the rusting of iron is as follows:[129]
- Cathode: 3 O2 + 6 H2O + 12 e− → 12 OH−
- Anode: 4 Fe → 4 Fe2+ + 8 e−; 4 Fe2+ → 4 Fe3+ + 4 e−
- Overall: 4 Fe + 3 O2 + 6 H2O → 4 Fe3+ + 12 OH− → 4 Fe(OH)3 or 4 FeO(OH) + 4 H2O
The electrolyte is usually iron(II) sulfate in urban areas (formed when atmospheric sulfur dioxide attacks iron), and salt particles in the atmosphere in seaside areas.[129]
Catalysts and reagents
Because Fe is inexpensive and nontoxic, much effort has been devoted to the development of Fe-based catalysts and reagents. Iron is however less common as a catalyst in commercial processes than more expensive metals.[130] In biology, Fe-containing enzymes are pervasive.[131]
Iron catalysts are traditionally used in the Haber–Bosch process for the production of ammonia and the Fischer–Tropsch process for conversion of carbon monoxide to hydrocarbons for fuels and lubricants.[132] Powdered iron in an acidic medium is used in the Bechamp reduction, the conversion of nitrobenzene to aniline.[133]
Iron compounds
Iron(III) oxide mixed with aluminium powder can be ignited to create a thermite reaction, used in welding large iron parts (like rails) and purifying ores. Iron(III) oxide and oxyhydroxide are used as reddish and ocher pigments.
Iron(III) chloride finds use in water purification and sewage treatment, in the dyeing of cloth, as a coloring agent in paints, as an additive in animal feed, and as an etchant for copper in the manufacture of printed circuit boards.[134] It can also be dissolved in alcohol to form tincture of iron, which is used as a medicine to stop bleeding in canaries.[135]
Iron(II) sulfate is used as a precursor to other iron compounds. It is also used to reduce chromate in cement. It is used to fortify foods and treat iron deficiency anemia. Iron(III) sulfate is used in settling minute sewage particles in tank water. Iron(II) chloride is used as a reducing flocculating agent, in the formation of iron complexes and magnetic iron oxides, and as a reducing agent in organic synthesis.[134]
Sodium nitroprusside is a drug used as a vasodilator. It is on the World Health Organization’s List of Essential Medicines.[136]
Biological and pathological role
Iron is required for life.[5][137][138] The iron–sulfur clusters are pervasive and include nitrogenase, the enzymes responsible for biological nitrogen fixation. Iron-containing proteins participate in transport, storage and used of oxygen.[5] Iron proteins are involved in electron transfer.[139]
Simplified structure of Heme b; in the protein additional ligand(s) are attached to Fe.
Examples of iron-containing proteins in higher organisms include hemoglobin, cytochrome (see high-valent iron), and catalase.[5][140] The average adult human contains about 0.005% body weight of iron, or about four grams, of which three quarters is in hemoglobin – a level that remains constant despite only about one milligram of iron being absorbed each day,[139] because the human body recycles its hemoglobin for the iron content.[141]
Microbial growth may be assisted by oxidation of iron(II) or by reduction of iron (III).[142]
Biochemistry
Iron acquisition poses a problem for aerobic organisms because ferric iron is poorly soluble near neutral pH. Thus, these organisms have developed means to absorb iron as complexes, sometimes taking up ferrous iron before oxidising it back to ferric iron.[5] In particular, bacteria have evolved very high-affinity sequestering agents called siderophores.[143][144][145]
After uptake in human cells, iron storage is precisely regulated.[5][146] A major component of this regulation is the protein transferrin, which binds iron ions absorbed from the duodenum and carries it in the blood to cells.[5][147] Transferrin contains Fe3+ in the middle of a distorted octahedron, bonded to one nitrogen, three oxygens and a chelating carbonate anion that traps the Fe3+ ion: it has such a high stability constant that it is very effective at taking up Fe3+ ions even from the most stable complexes. At the bone marrow, transferrin is reduced from Fe3+ and Fe2+ and stored as ferritin to be incorporated into hemoglobin.[139]
The most commonly known and studied bioinorganic iron compounds (biological iron molecules) are the heme proteins: examples are hemoglobin, myoglobin, and cytochrome P450.[5] These compounds participate in transporting gases, building enzymes, and transferring electrons.[139] Metalloproteins are a group of proteins with metal ion cofactors. Some examples of iron metalloproteins are ferritin and rubredoxin.[139] Many enzymes vital to life contain iron, such as catalase,[148] lipoxygenases,[149] and IRE-BP.[150]
Hemoglobin is an oxygen carrier that occurs in red blood cells and contributes their color, transporting oxygen in the arteries from the lungs to the muscles where it is transferred to myoglobin, which stores it until it is needed for the metabolic oxidation of glucose, generating energy.[5] Here the hemoglobin binds to carbon dioxide, produced when glucose is oxidized, which is transported through the veins by hemoglobin (predominantly as bicarbonate anions) back to the lungs where it is exhaled.[139] In hemoglobin, the iron is in one of four heme groups and has six possible coordination sites; four are occupied by nitrogen atoms in a porphyrin ring, the fifth by an imidazole nitrogen in a histidine residue of one of the protein chains attached to the heme group, and the sixth is reserved for the oxygen molecule it can reversibly bind to.[139] When hemoglobin is not attached to oxygen (and is then called deoxyhemoglobin), the Fe2+ ion at the center of the heme group (in the hydrophobic protein interior) is in a high-spin configuration. It is thus too large to fit inside the porphyrin ring, which bends instead into a dome with the Fe2+ ion about 55 picometers above it. In this configuration, the sixth coordination site reserved for the oxygen is blocked by another histidine residue.[139]
When deoxyhemoglobin picks up an oxygen molecule, this histidine residue moves away and returns once the oxygen is securely attached to form a hydrogen bond with it. This results in the Fe2+ ion switching to a low-spin configuration, resulting in a 20% decrease in ionic radius so that now it can fit into the porphyrin ring, which becomes planar.[139] (Additionally, this hydrogen bonding results in the tilting of the oxygen molecule, resulting in a Fe–O–O bond angle of around 120° that avoids the formation of Fe–O–Fe or Fe–O2–Fe bridges that would lead to electron transfer, the oxidation of Fe2+ to Fe3+, and the destruction of hemoglobin.) This results in a movement of all the protein chains that leads to the other subunits of hemoglobin changing shape to a form with larger oxygen affinity. Thus, when deoxyhemoglobin takes up oxygen, its affinity for more oxygen increases, and vice versa.[139] Myoglobin, on the other hand, contains only one heme group and hence this cooperative effect cannot occur. Thus, while hemoglobin is almost saturated with oxygen in the high partial pressures of oxygen found in the lungs, its affinity for oxygen is much lower than that of myoglobin, which oxygenates even at low partial pressures of oxygen found in muscle tissue.[139] As described by the Bohr effect (named after Christian Bohr, the father of Niels Bohr), the oxygen affinity of hemoglobin diminishes in the presence of carbon dioxide.[139]
Carbon monoxide and phosphorus trifluoride are poisonous to humans because they bind to hemoglobin similarly to oxygen, but with much more strength, so that oxygen can no longer be transported throughout the body. Hemoglobin bound to carbon monoxide is known as carboxyhemoglobin. This effect also plays a minor role in the toxicity of cyanide, but there the major effect is by far its interference with the proper functioning of the electron transport protein cytochrome a.[139] The cytochrome proteins also involve heme groups and are involved in the metabolic oxidation of glucose by oxygen. The sixth coordination site is then occupied by either another imidazole nitrogen or a methionine sulfur, so that these proteins are largely inert to oxygen – with the exception of cytochrome a, which bonds directly to oxygen and thus is very easily poisoned by cyanide.[139] Here, the electron transfer takes place as the iron remains in low spin but changes between the +2 and +3 oxidation states. Since the reduction potential of each step is slightly greater than the previous one, the energy is released step-by-step and can thus be stored in adenosine triphosphate. Cytochrome a is slightly distinct, as it occurs at the mitochondrial membrane, binds directly to oxygen, and transports protons as well as electrons, as follows:[139]
- 4 Cytc2+ + O2 + 8H+
inside → 4 Cytc3+ + 2 H2O + 4H+
outside
Although the heme proteins are the most important class of iron-containing proteins, the iron–sulfur proteins are also very important, being involved in electron transfer, which is possible since iron can exist stably in either the +2 or +3 oxidation states. These have one, two, four, or eight iron atoms that are each approximately tetrahedrally coordinated to four sulfur atoms; because of this tetrahedral coordination, they always have high-spin iron. The simplest of such compounds is rubredoxin, which has only one iron atom coordinated to four sulfur atoms from cysteine residues in the surrounding peptide chains. Another important class of iron–sulfur proteins is the ferredoxins, which have multiple iron atoms. Transferrin does not belong to either of these classes.[139]
The ability of sea mussels to maintain their grip on rocks in the ocean is facilitated by their use of organometallic iron-based bonds in their protein-rich cuticles. Based on synthetic replicas, the presence of iron in these structures increased elastic modulus 770 times, tensile strength 58 times, and toughness 92 times. The amount of stress required to permanently damage them increased 76 times.[152]
Nutrition
Diet
Iron is pervasive, but particularly rich sources of dietary iron include red meat, oysters, beans, poultry, fish, leaf vegetables, watercress, tofu, and blackstrap molasses.[5] Bread and breakfast cereals are sometimes specifically fortified with iron.[5][153]
Iron provided by dietary supplements is often found as iron(II) fumarate, although iron(II) sulfate is cheaper and is absorbed equally well.[134] Elemental iron, or reduced iron, despite being absorbed at only one-third to two-thirds the efficiency (relative to iron sulfate),[154] is often added to foods such as breakfast cereals or enriched wheat flour. Iron is most available to the body when chelated to amino acids[155] and is also available for use as a common iron supplement. Glycine, the least expensive amino acid, is most often used to produce iron glycinate supplements.[156]
Dietary recommendations
The U.S. Institute of Medicine (IOM) updated Estimated Average Requirements (EARs) and Recommended Dietary Allowances (RDAs) for iron in 2001.[5] The current EAR for iron for women ages 14–18 is 7.9 mg/day, 8.1 for ages 19–50 and 5.0 thereafter (post menopause). For men the EAR is 6.0 mg/day for ages 19 and up. The RDA is 15.0 mg/day for women ages 15–18, 18.0 for 19–50 and 8.0 thereafter. For men, 8.0 mg/day for ages 19 and up. RDAs are higher than EARs so as to identify amounts that will cover people with higher than average requirements. RDA for pregnancy is 27 mg/day and, for lactation, 9 mg/day.[5] For children ages 1–3 years 7 mg/day, 10 for ages 4–8 and 8 for ages 9–13. As for safety, the IOM also sets Tolerable upper intake levels (ULs) for vitamins and minerals when evidence is sufficient. In the case of iron the UL is set at 45 mg/day. Collectively the EARs, RDAs and ULs are referred to as Dietary Reference Intakes.[157]
The European Food Safety Authority (EFSA) refers to the collective set of information as Dietary Reference Values, with Population Reference Intake (PRI) instead of RDA, and Average Requirement instead of EAR. AI and UL defined the same as in United States. For women the PRI is 13 mg/day ages 15–17 years, 16 mg/day for women ages 18 and up who are premenopausal and 11 mg/day postmenopausal. For pregnancy and lactation, 16 mg/day. For men the PRI is 11 mg/day ages 15 and older. For children ages 1 to 14 the PRI increases from 7 to 11 mg/day. The PRIs are higher than the U.S. RDAs, with the exception of pregnancy.[158] The EFSA reviewed the same safety question did not establish a UL.[159]
Infants may require iron supplements if they are bottle-fed cow’s milk.[160] Frequent blood donors are at risk of low iron levels and are often advised to supplement their iron intake.[161]
For U.S. food and dietary supplement labeling purposes the amount in a serving is expressed as a percent of Daily Value (%DV). For iron labeling purposes 100% of the Daily Value was 18 mg, and as of May 27, 2016 remained unchanged at 18 mg.[162][163] A table of the old and new adult daily values is provided at Reference Daily Intake.
Deficiency
Iron deficiency is the most common nutritional deficiency in the world.[5][164][165][166] When loss of iron is not adequately compensated by adequate dietary iron intake, a state of latent iron deficiency occurs, which over time leads to iron-deficiency anemia if left untreated, which is characterised by an insufficient number of red blood cells and an insufficient amount of hemoglobin.[167] Children, pre-menopausal women (women of child-bearing age), and people with poor diet are most susceptible to the disease. Most cases of iron-deficiency anemia are mild, but if not treated can cause problems like fast or irregular heartbeat, complications during pregnancy, and delayed growth in infants and children.[168]
Excess
Iron uptake is tightly regulated by the human body, which has no regulated physiological means of excreting iron. Only small amounts of iron are lost daily due to mucosal and skin epithelial cell sloughing, so control of iron levels is primarily accomplished by regulating uptake.[169] Regulation of iron uptake is impaired in some people as a result of a genetic defect that maps to the HLA-H gene region on chromosome 6 and leads to abnormally low levels of hepcidin, a key regulator of the entry of iron into the circulatory system in mammals.[170] In these people, excessive iron intake can result in iron overload disorders, known medically as hemochromatosis.[5] Many people have an undiagnosed genetic susceptibility to iron overload, and are not aware of a family history of the problem. For this reason, people should not take iron supplements unless they suffer from iron deficiency and have consulted a doctor. Hemochromatosis is estimated to be the cause of 0.3 to 0.8% of all metabolic diseases of Caucasians.[171]
Overdoses of ingested iron can cause excessive levels of free iron in the blood. High blood levels of free ferrous iron react with peroxides to produce highly reactive free radicals that can damage DNA, proteins, lipids, and other cellular components. Iron toxicity occurs when the cell contains free iron, which generally occurs when iron levels exceed the availability of transferrin to bind the iron. Damage to the cells of the gastrointestinal tract can also prevent them from regulating iron absorption, leading to further increases in blood levels. Iron typically damages cells in the heart, liver and elsewhere, causing adverse effects that include coma, metabolic acidosis, shock, liver failure, coagulopathy, long-term organ damage, and even death.[172] Humans experience iron toxicity when the iron exceeds 20 milligrams for every kilogram of body mass; 60 milligrams per kilogram is considered a lethal dose.[173] Overconsumption of iron, often the result of children eating large quantities of ferrous sulfate tablets intended for adult consumption, is one of the most common toxicological causes of death in children under six.[173] The Dietary Reference Intake (DRI) sets the Tolerable Upper Intake Level (UL) for adults at 45 mg/day. For children under fourteen years old the UL is 40 mg/day.[174]
The medical management of iron toxicity is complicated, and can include use of a specific chelating agent called deferoxamine to bind and expel excess iron from the body.[172][175][176]
ADHD
Some research has suggested that low thalamic iron levels may play a role in the pathophysiology of ADHD.[177] Some researchers have found that iron supplementation can be effective especially in the inattentive subtype of the disorder.[178] One study also showed that iron may be able to decrease the risk of cardiovascular events during treatment with ADHD drugs.[179]
Some researchers in the 2000s suggested a link between low levels of iron in the blood and ADHD. A 2012 study found no such correlation.[180]
Cancer
The role of iron in cancer defense can be described as a «double-edged sword» because of its pervasive presence in non-pathological processes.[181] People having chemotherapy may develop iron deficiency and anemia, for which intravenous iron therapy is used to restore iron levels.[182] Iron overload, which may occur from high consumption of red meat,[5] may initiate tumor growth and increase susceptibility to cancer onset,[182] particularly for colorectal cancer.[5]
Marine systems
Iron plays an essential role in marine systems and can act as a limiting nutrient for planktonic activity.[183] Because of this, too much of a decrease in iron may lead to a decrease in growth rates in phytoplanktonic organisms such as diatoms.[184] Iron can also be oxidized by marine microbes under conditions that are high in iron and low in oxygen.[185]
Iron can enter marine systems through adjoining rivers and directly from the atmosphere. Once iron enters the ocean, it can be distributed throughout the water column through ocean mixing and through recycling on the cellular level.[186] In the arctic, sea ice plays a major role in the store and distribution of iron in the ocean, depleting oceanic iron as it freezes in the winter and releasing it back into the water when thawing occurs in the summer.[187] The iron cycle can fluctuate the forms of iron from aqueous to particle forms altering the availability of iron to primary producers.[188] Increased light and warmth increases the amount of iron that is in forms that are usable by primary producers.[189]
See also
- El Mutún in Bolivia, where 10% of the world’s accessible iron ore is located
- Iron and steel industry
- Iron cycle
- Iron nanoparticle
- Iron–platinum nanoparticle
- Iron fertilization – proposed fertilization of oceans to stimulate phytoplankton growth
- Iron-oxidizing bacteria
- List of countries by iron production
- Pelletising – process of creation of iron ore pellets
- Rustproof iron
- Steel
References
- ^ «Standard Atomic Weights: Iron». CIAAW. 1993.
- ^ Ram, R. S.; Bernath, P. F. (2003). «Fourier transform emission spectroscopy of the g4Δ–a4Δ system of FeCl». Journal of Molecular Spectroscopy. 221 (2): 261. Bibcode:2003JMoSp.221..261R. doi:10.1016/S0022-2852(03)00225-X.
- ^ Demazeau, G.; Buffat, B.; Pouchard, M.; Hagenmuller, P. (1982). «Recent developments in the field of high oxidation states of transition elements in oxides stabilization of six-coordinated Iron(V)». Zeitschrift für anorganische und allgemeine Chemie. 491: 60–66. doi:10.1002/zaac.19824910109.
- ^ Lu, J.; Jian, J.; Huang, W.; Lin, H.; Li, J; Zhou, M. (2016). «Experimental and theoretical identification of the Fe(VII) oxidation state in FeO4−«. Physical Chemistry Chemical Physics. 18 (45): 31125–31131. Bibcode:2016PCCP…1831125L. doi:10.1039/C6CP06753K. PMID 27812577.
- ^ a b c d e f g h i j k l m n o p q «Iron». Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis, Oregon. April 2016. Retrieved 6 March 2018.
- ^ a b c d e f g h Greenwood & Earnshaw 1997, pp. 1075–79.
- ^ Hirose, K., Tateno, S. (2010). «The Structure of Iron in Earth’s Inner Core». Science. American Association for the Advancement of Science. 330 (6002): 359–361. Bibcode:2010Sci…330..359T. doi:10.1126/science.1194662. PMID 20947762. S2CID 206528628.
- ^ Chamati, Gaminchev (2014). «Dynamic stability of Fe under high pressure». Journal of Physics. IOP Publishing. 558 (1): 012013. Bibcode:2014JPhCS.558a2013G. doi:10.1088/1742-6596/558/1/012013.
- ^ Boehler, Reinhard (2000). «High-pressure experiments and the phase diagram of lower mantle and core materials». Reviews of Geophysics. American Geophysical Union. 38 (2): 221–45. Bibcode:2000RvGeo..38..221B. doi:10.1029/1998RG000053. S2CID 33458168.
- ^ Stixrude, Lars; Wasserman, Evgeny; Cohen, Ronald E. (10 November 1997). «Composition and temperature of Earth’s inner core». Journal of Geophysical Research: Solid Earth. 102 (B11): 24729–39. Bibcode:1997JGR…10224729S. doi:10.1029/97JB02125.
- ^ Greenwood & Earnshaw 1997, p. 1116.
- ^ a b c d e f Greenwood & Earnshaw 1997, pp. 1074–75.
- ^ Boehler, Reinhard; Ross, M. (2007). «Properties of Rocks and Minerals_High-Pressure Melting». Mineral Physics. Treatise on Geophysics. Vol. 2. Elsevier. pp. 527–41. doi:10.1016/B978-044452748-6.00047-X. ISBN 9780444527486.
- ^ Steinmetz, Charles (1917). «fig. 42». Theory and Calculation of Electric Circuits. McGraw-Hill.
- ^ a b Cullity; C. D. Graham (2008). Introduction to Magnetic Materials, 2nd. New York: Wiley–IEEE. p. 116. ISBN 978-0-471-47741-9.
- ^ a b Bramfitt, B.L.; Benscoter, Arlan O. (2002). «The Iron Carbon Phase Diagram». Metallographer’s guide: practice and procedures for irons and steels. ASM International. pp. 24–28. ISBN 978-0-87170-748-2.
- ^ a b Audi, Georges; Bersillon, Olivier; Blachot, Jean; Wapstra, Aaldert Hendrik (2003), «The NUBASE evaluation of nuclear and decay properties», Nuclear Physics A, 729: 3–128, Bibcode:2003NuPhA.729….3A, doi:10.1016/j.nuclphysa.2003.11.001
- ^ Rugel, G.; Faestermann, T.; Knie, K.; Korschinek, G.; Poutivtsev, M.; Schumann, D.; Kivel, N.; Günther-Leopold, I.; Weinreich, R.; Wohlmuther, M. (2009). «New Measurement of the 60Fe Half-Life». Physical Review Letters. 103 (7): 072502. Bibcode:2009PhRvL.103g2502R. doi:10.1103/PhysRevLett.103.072502. PMID 19792637.
- ^ Dauphas, N.; Rouxel, O. (2006). «Mass spectrometry and natural variations of iron isotopes» (PDF). Mass Spectrometry Reviews. 25 (4): 515–50. Bibcode:2006MSRv…25..515D. doi:10.1002/mas.20078. PMID 16463281. Archived from the original (PDF) on 10 June 2010.
- ^ Mostefaoui, S.; Lugmair, G.W.; Hoppe, P.; El Goresy, A. (2004). «Evidence for live 60Fe in meteorites». New Astronomy Reviews. 48 (1–4): 155–59. Bibcode:2004NewAR..48..155M. doi:10.1016/j.newar.2003.11.022.
- ^ Fewell, M. P. (1995). «The atomic nuclide with the highest mean binding energy». American Journal of Physics. 63 (7): 653. Bibcode:1995AmJPh..63..653F. doi:10.1119/1.17828.
- ^ a b c Greenwood & Earnshaw 1997, p. 12.
- ^ Woosley, S.; Janka, T. (2006). «The physics of core collapse supernovae». Nature Physics. 1 (3): 147–54. arXiv:astro-ph/0601261. Bibcode:2005NatPh…1..147W. doi:10.1038/nphys172. S2CID 118974639.
- ^ McDonald, I.; Sloan, G. C.; Zijlstra, A. A.; Matsunaga, N.; Matsuura, M.; Kraemer, K. E.; Bernard-Salas, J.; Markwick, A. J. (2010). «Rusty Old Stars: A Source of the Missing Interstellar Iron?». The Astrophysical Journal Letters. 717 (2): L92–L97. arXiv:1005.3489. Bibcode:2010ApJ…717L..92M. doi:10.1088/2041-8205/717/2/L92. S2CID 14437704.
- ^ Bautista, Manuel A.; Pradhan, Anil K. (1995). «Iron and Nickel Abundances in H~II Regions and Supernova Remnants». Bulletin of the American Astronomical Society. 27: 865. Bibcode:1995AAS…186.3707B.
- ^ Dyson, Freeman J. (1979). «Time without end: Physics and biology in an open universe». Reviews of Modern Physics. 51 (3): 447–60. Bibcode:1979RvMP…51..447D. doi:10.1103/RevModPhys.51.447.
- ^ Aron, Jacob. «Supernova space bullets could have seeded Earth’s iron core». New Scientist. Retrieved 2 October 2020.
- ^ Croswell, Ken. «Iron in the Fire: The Little-Star Supernovae That Could». Scientific American. Retrieved 3 January 2021.
- ^ Buchwald, V F (1992). «On the Use of Iron by the Eskimos in Greenland». Materials Characterization. 29 (2): 139–176. doi:10.1016/1044-5803(92)90112-U.
- ^ Emiliani, Cesare (1992). Planet earth: cosmology, geology, and the evolution of life and environment. Cambridge University Press. p. 152. Bibcode:1992pecg.book…..E. ISBN 978-0-521-40949-0.
- ^ Pernet-Fisher, J.; Day, J.M.D.; Howarth, G.H.; Ryabov, V.V.; Taylor, L.A. (2017). «Atmospheric outgassing and native-iron formation during carbonaceous sediment–basalt melt interactions». Earth and Planetary Science Letters. 460: 201–212. Bibcode:2017E&PSL.460..201P. doi:10.1016/j.epsl.2016.12.022.
- ^ Stark, Anne M. (20 September 2007) Researchers locate mantle’s spin transition zone, leading to clues about earth’s structure. Lawrence Livermore National Laboratory
- ^ Ferropericlase. Mindat.org
- ^ Murakami, M.; Ohishi Y.; Hirao N.; Hirose K. (2012). «A perovskitic lower mantle inferred from high-pressure, high-temperature sound velocity data». Nature. 485 (7396): 90–94. Bibcode:2012Natur.485…90M. doi:10.1038/nature11004. PMID 22552097. S2CID 4387193.
- ^ Sharp, T. (27 November 2014). «Bridgmanite – named at last». Science. 346 (6213): 1057–58. Bibcode:2014Sci…346.1057S. doi:10.1126/science.1261887. PMID 25430755. S2CID 206563252.
- ^ Kong, L. T.; Li, J. F.; Shi, Q. W.; Huang, H. J.; Zhao, K. (6 March 2012). «Dynamical stability of iron under high-temperature and high-pressure conditions». EPL. 97 (5): 56004p1–56004p5. Bibcode:2012EL…..9756004K. doi:10.1209/0295-5075/97/56004. S2CID 121861429.
- ^ Gaminchev, K. G.; Chamati, H. (3 December 2014). «Dynamic stability of Fe under high pressure». J. Phys. 558 (1): 012013(1–7). Bibcode:2014JPhCS.558a2013G. doi:10.1088/1742-6596/558/1/012013.
- ^ Morgan, John W. & Anders, Edward (1980). «Chemical composition of Earth, Venus, and Mercury». Proc. Natl. Acad. Sci. 77 (12): 6973–77. Bibcode:1980PNAS…77.6973M. doi:10.1073/pnas.77.12.6973. PMC 350422. PMID 16592930.
- ^ «Pyrrhotite». Mindat.org. Retrieved 7 July 2009.
- ^ Klein, Cornelis and Cornelius S. Hurlbut, Jr. (1985) Manual of Mineralogy, Wiley, 20th ed, pp. 278–79 ISBN 0-471-80580-7
- ^ a b Greenwood & Earnshaw 1997, p. 1071.
- ^ Lyons, T. W.; Reinhard, C. T. (2009). «Early Earth: Oxygen for heavy-metal fans». Nature. 461 (7261): 179–181. Bibcode:2009Natur.461..179L. doi:10.1038/461179a. PMID 19741692. S2CID 205049360.
- ^ Cloud, P. (1973). «Paleoecological Significance of the Banded Iron-Formation». Economic Geology. 68 (7): 1135–43. doi:10.2113/gsecongeo.68.7.1135.
- ^ Dickinson, Robert E. (1964). Germany: A regional and economic geography (2nd ed.). London: Methuen.
- ^ Naturwerksteine in Baden-Württemberg. Landesamt für Geologie, Rohstoffe und Bergbau, Baden-Württemberg
- ^ «Tales From The Riverbank». Minerva Stone Conservation. Archived from the original on 28 September 2015. Retrieved 22 September 2015.
- ^ Klingelhöfer, G.; Morris, R. V.; Souza, P. A.; Rodionov, D.; Schröder, C. (2007). «Two earth years of Mössbauer studies of the surface of Mars with MIMOS II». Hyperfine Interactions. 170 (1–3): 169–77. Bibcode:2006HyInt.170..169K. doi:10.1007/s10751-007-9508-5. S2CID 98227499.
- ^ Winderlich, R.; Peter, W. (1954). Lehrbuch der Chemie für Höhere Lehranstalten : Einheitsausgabe für Unter- und Oberstufe (in German). Wiesbaden. p. 75. ISBN 978-3-663-04370-6. OCLC 913701506.
- ^ Bertau, Martin (2013). Industrielle Anorganische Chemie (in German). Weinheim: Wiley-VCH. p. 696. ISBN 978-3-527-64956-3. OCLC 855858511.
- ^ Metal Stocks in Society: Scientific synthesis, 2010, International Resource Panel, UNEP
- ^ Stoll, Heather (17 February 2020). «30 years of the iron hypothesis of ice ages». Nature. Springer Science and Business Media LLC. 578 (7795): 370–371. doi:10.1038/d41586-020-00393-x. ISSN 0028-0836.
- ^ Greenwood & Earnshaw 1997, p. 905.
- ^ a b Greenwood & Earnshaw 1997, p. 1070.
- ^ Lu, Jun-Bo; Jian, Jiwen; Huang, Wei; Lin, Hailu; Li, Jun; Zhou, Mingfei (16 November 2016). «Experimental and theoretical identification of the Fe(VII) oxidation state in FeO4−«. Phys. Chem. Chem. Phys. 18 (45): 31125–31131. Bibcode:2016PCCP…1831125L. doi:10.1039/c6cp06753k. PMID 27812577.
- ^ Nam, Wonwoo (2007). «High-Valent Iron(IV)–Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions» (PDF). Accounts of Chemical Research. 40 (7): 522–531. doi:10.1021/ar700027f. PMID 17469792. Archived from the original (PDF) on 15 June 2021. Retrieved 22 February 2022.
- ^ a b c d e f Holleman, Arnold F.; Wiberg, Egon; Wiberg, Nils (1985). «Iron». Lehrbuch der Anorganischen Chemie (in German) (91–100 ed.). Walter de Gruyter. pp. 1125–46. ISBN 3-11-007511-3.
- ^ Reiff, William Michael; Long, Gary J. (1984). «Mössbauer Spectroscopy and the Coordination Chemistry of Iron». Mössbauer spectroscopy applied to inorganic chemistry. Springer. pp. 245–83. ISBN 978-0-306-41647-7.
- ^ Ware, Mike (1999). «An introduction in monochrome». Cyanotype: the history, science and art of photographic printing in Prussian blue. NMSI Trading Ltd. pp. 11–19. ISBN 978-1-900747-07-3.
- ^ Gmelin, Leopold (1852). «Mercury and Iron». Hand-book of chemistry. Vol. 6. Cavendish Society. pp. 128–29.
- ^ a b c Greenwood & Earnshaw 1997, p. 1079.
- ^ a b c d Greenwood & Earnshaw 1997, pp. 1082–84.
- ^ Siegfried Pohl, Ulrich Bierbach, Wolfgang Saak; «FeI3SC(NMe2)2, a Neutral Thiourea Complex of Iron(III) Iodide», Angewandte Chemie International Edition in English (1989) 28 (6), 776-777. https://doi.org/10.1002/anie.198907761
- ^ Nicholas A. Barnes, Stephen M.Godfrey, Nicholas Ho, Charles A.McAuliffe, Robin G.Pritchard; «Facile synthesis of a rare example of an iron(III) iodide complex, [FeI3(AsMe3)2], from the reaction of Me3AsI2 with unactivated iron powder», Polyhedron (2013) 55, 67-72. https://doi.org/10.1016/j.poly.2013.02.066
- ^ a b c d Greenwood & Earnshaw 1997, pp. 1088–91.
- ^ a b Greenwood & Earnshaw 1997, pp. 1091–97.
- ^ Clausen, C.A.; Good, M.L. (1968). «Stabilization of the hexachloroferrate(III) anion by the methylammonium cation». Inorganic Chemistry. 7 (12): 2662–63. doi:10.1021/ic50070a047.
- ^ James, B.D.; Bakalova, M.; Lieseganga, J.; Reiff, W.M.; Hockless, D.C.R.; Skelton, B.W.; White, A.H. (1996). «The hexachloroferrate(III) anion stabilized in hydrogen bonded packing arrangements. A comparison of the X-ray crystal structures and low temperature magnetism of tetrakis(methylammonium) hexachloroferrate(III) chloride (I) and tetrakis(hexamethylenediammonium) hexachloroferrate(III) tetrachloroferrate(III) tetrachloride (II)«. Inorganica Chimica Acta. 247 (2): 169–74. doi:10.1016/0020-1693(95)04955-X.
- ^ Giannoccaro, P.; Sacco, A. (1977). Bis[ethylenebis(diphenylphosphine)]-Hydridoiron Complexes. Inorg. Synth. Inorganic Syntheses. Vol. 17. pp. 69–72. doi:10.1002/9780470132487.ch19. ISBN 978-0-470-13248-7.
- ^ Lee, J.; Jung, G.; Lee, S.W. (1998). «Structure of trans-chlorohydridobis(diphenylphosphinoethane)iron(II)». Bull. Korean Chem. Soc. 19 (2): 267–69. doi:10.1007/BF02698412. S2CID 35665289.
- ^ Echigo, Takuya; Kimata, Mitsuyoshi (2008). «Single-crystal X-ray diffraction and spectroscopic studies on humboldtine and lindbergite: weak Jahn–Teller effect of Fe2+ ion». Phys. Chem. Minerals. 35 (8): 467–75. Bibcode:2008PCM….35..467E. doi:10.1007/s00269-008-0241-7. S2CID 98739882.
- ^ Greenwood, Norman N.; Earnshaw, Alan (1984). Chemistry of the Elements. Oxford: Pergamon Press. pp. 1282–86. ISBN 978-0-08-022057-4..
- ^ Kealy, T.J.; Pauson, P.L. (1951). «A New Type of Organo-Iron Compound». Nature. 168 (4285): 1039–40. Bibcode:1951Natur.168.1039K. doi:10.1038/1681039b0. S2CID 4181383.
- ^ Miller, S. A.; Tebboth, J. A.; Tremaine, J. F. (1952). «114. Dicyclopentadienyliron». J. Chem. Soc.: 632–635. doi:10.1039/JR9520000632.
- ^ Wilkinson, G.; Rosenblum, M.; Whiting, M. C.; Woodward, R. B. (1952). «The Structure of Iron Bis-Cyclopentadienyl». J. Am. Chem. Soc. 74 (8): 2125–2126. doi:10.1021/ja01128a527.
- ^ Okuda, Jun (28 December 2016). «Ferrocene – 65 Years After». European Journal of Inorganic Chemistry. 2017 (2): 217–219. doi:10.1002/ejic.201601323. ISSN 1434-1948.
- ^ Greenwood & Earnshaw 1997, p. 1104.
- ^ Bullock, R.M. (11 September 2007). «An Iron Catalyst for Ketone Hydrogenations under Mild Conditions». Angew. Chem. Int. Ed. 46 (39): 7360–63. doi:10.1002/anie.200703053. PMID 17847139.
- ^ Weeks 1968, p. 4.
- ^ a b Weeks 1968, p. 29.
- ^ a b c Weeks 1968, p. 31.
- ^ Bjorkman, Judith Kingston (1973). «Meteors and Meteorites in the ancient Near East». Meteoritics. 8 (2): 91–132. Bibcode:1973Metic…8…91B. doi:10.1111/j.1945-5100.1973.tb00146.x.
- ^ Comelli, Daniela; d’Orazio, Massimo; Folco, Luigi; El-Halwagy, Mahmud; Frizzi, Tommaso; Alberti, Roberto; Capogrosso, Valentina; Elnaggar, Abdelrazek; Hassan, Hala; Nevin, Austin; Porcelli, Franco; Rashed, Mohamed G; Valentini, Gianluca (2016). «The meteoritic origin of Tutankhamun’s iron dagger blade». Meteoritics & Planetary Science. 51 (7): 1301–09. Bibcode:2016M&PS…51.1301C. doi:10.1111/maps.12664.
- ^ Walsh, Declan (2 June 2016). «King Tut’s Dagger Made of ‘Iron From the Sky,’ Researchers Say». The New York Times. Archived from the original on 3 January 2022. Retrieved 4 June 2016.
the blade’s composition of iron, nickel and cobalt was an approximate match for a meteorite that landed in northern Egypt. The result «strongly suggests an extraterrestrial origin»
- ^ Ure, Andrew (1843). Technisches wörterbuch oder Handbuch der Gewerbskunde … : Bearb. nach Dr. Andrew Ure’s Dictionary of arts, manufactures and mines (in German). G. Haase. p. 492.
- ^ a b c d Weeks 1968, p. 32.
- ^ McNutt, Paula (1990 1). The Forging of Israel: Iron Technology, Symbolism and Tradition in Ancient Society. A&C Black.
- ^ Tewari, Rakesh. «The origins of Iron Working in India: New evidence from the Central Ganga plain and the Eastern Vindhyas» (PDF). State Archaeological Department. Retrieved 23 May 2010.
- ^ Photos, E. (1989). «The Question of Meteoritic versus Smelted Nickel-Rich Iron: Archaeological Evidence and Experimental Results». World Archaeology. Taylor & Francis, Ltd. 20 (3): 403–21. doi:10.1080/00438243.1989.9980081. JSTOR 124562.
- ^ Muhly, James D. (2003). «Metalworking/Mining in the Levant». In Lake, Richard Winona (ed.). Near Eastern Archaeology IN: Eisenbrauns. Vol. 180. pp. 174–83.
- ^ Witzel, Michael (2001), «Autochthonous Aryans? The Evidence from Old Indian and Iranian Texts», in Electronic Journal of Vedic Studies (EJVS) 7-3, pp. 1–93
- ^ Weeks, p. 33, quoting Cline, Walter (1937) «Mining and Metallurgy in Negro Africa,» George Banta Publishing Co., Menasha, Wis., pp. 17–23.
- ^ Riederer, Josef; Wartke, Ralf-B. (2009) «Iron», Cancik, Hubert; Schneider, Helmuth (eds.): Brill’s New Pauly, Brill.
- ^ Sawyer, Ralph D. and Sawyer, Mei-chün (1993). The Seven Military Classics of Ancient China. Boulder: Westview. ISBN 0-465-00304-4. p. 10.
- ^ Pigott, Vincent C. (1999). The Archaeometallurgy of the Asian Old World. Philadelphia: University of Pennsylvania Museum of Archaeology and Anthropology. ISBN 0-924171-34-0, p. 8.
- ^ Golas, Peter J. (1999). Science and Civilisation in China: Volume 5, Chemistry and Chemical Technology, Part 13, Mining. Cambridge University Press. p. 152. ISBN 978-0-521-58000-7.
earliest blast furnace discovered in China from about the first century AD
- ^ Pigott, Vincent C. (1999). The Archaeometallurgy of the Asian Old World. Philadelphia: University of Pennsylvania Museum of Archaeology and Anthropology. ISBN 0-924171-34-0, p. 191.
- ^ The Coming of the Ages of Steel. Brill Archive. 1961. p. 54.
- ^ Mott, R.A (2014). «Dry and Wet Puddling». Transactions of the Newcomen Society. 49: 156–57. doi:10.1179/tns.1977.011.
- ^ Wagner, Donald B. (2003). «Chinese blast furnaces from the 10th to the 14th century» (PDF). Historical Metallurgy. 37 (1): 25–37. Archived from the original (PDF) on 7 January 2018. Retrieved 7 January 2018. originally published in Wagner, Donald B. (2001). «Chinese blast furnaces from the 10th to the 14th century». West Asian Science, Technology, and Medicine. 18: 41–74. doi:10.1163/26669323-01801008.
- ^ Giannichedda, Enrico (2007): «Metal production in Late Antiquity», in Technology in Transition AD 300–650 Lavan, L.; Zanini, E. and Sarantis, A.(eds.), Brill, Leiden; ISBN 90-04-16549-5, p. 200.
- ^ a b c d e f g Biddle, Verne; Parker, Gregory. Chemistry, Precision and Design. A Beka Book, Inc.
- ^ Wagner, Donald B. (1993). Iron and Steel in Ancient China. Brill. pp. 335–340. ISBN 978-90-04-09632-5.
- ^ a b c d Greenwood & Earnshaw 1997, p. 1072.
- ^ Schivelbusch, G. (1986) The Railway Journey: Industrialization and Perception of Time and Space in the 19th Century. Oxford: Berg.
- ^ Spoerl, Joseph S. A Brief History of Iron and Steel Production Archived 2 June 2010 at the Wayback Machine. Saint Anselm College
- ^ Enghag, Per (8 January 2008). Encyclopedia of the Elements: Technical Data – History – Processing – Applications. pp. 190–91. ISBN 978-3-527-61234-5.
- ^ Whitaker, Robert D (1975). «An historical note on the conservation of mass». Journal of Chemical Education. 52 (10): 658. Bibcode:1975JChEd..52..658W. doi:10.1021/ed052p658.
- ^ Fontenrose, Joseph (1974). «Work, Justice, and Hesiod’s Five Ages». Classical Philology. 69 (1): 1–16. doi:10.1086/366027. JSTOR 268960. S2CID 161808359.
- ^ Schmidt, Eva (1981) Der preußische Eisenkunstguss. (Art of Prussian cast iron) Technik, Geschichte, Werke, Künstler. Verlag Mann, Berlin, ISBN 3-7861-1130-8
- ^ Lux, H. (1963) «Metallic Iron» in Handbook of Preparative Inorganic Chemistry, 2nd Ed. G. Brauer (ed.), Academic Press, NY. Vol. 2. pp. 1490–91.
- ^ Steel Statistical Yearbook 2010. World Steel Association
- ^ a b c d e f g Greenwood & Earnshaw 1997, p. 1073.
- ^ Song Yingxing (1637): The Tiangong Kaiwu encyclopedia.
- ^ Wang, Peng; Ryberg, Morten; Yang, Yi; Feng, Kuishuang; Kara, Sami; Hauschild, Michael; Chen, Wei-Qiang (6 April 2021). «Efficiency stagnation in global steel production urges joint supply- and demand-side mitigation efforts». Nature Communications. 12 (1): 2066. Bibcode:2021NatCo..12.2066W. doi:10.1038/s41467-021-22245-6. ISSN 2041-1723. PMC 8024266. PMID 33824307.
- ^ Verhoeven, J.D. (1975) Fundamentals of Physical Metallurgy, Wiley, New York, p. 326
- ^ Greenwood & Earnshaw 1997, pp. 1070–71.
- ^ a b Kohl, Walter H. (1995). Handbook of materials and techniques for vacuum devices. Springer. pp. 164–67. ISBN 1-56396-387-6.
- ^ a b Kuhn, Howard; Medlin, Dana; et al., eds. (2000). ASM Handbook – Mechanical Testing and Evaluation (PDF). Vol. 8. ASM International. p. 275. ISBN 0-87170-389-0. Archived from the original (PDF) on 9 February 2019. Retrieved 22 February 2022.
- ^ «Hardness Conversion Chart». Maryland Metrics. Archived from the original on 18 June 2015. Retrieved 23 May 2010.
- ^ Takaji, Kusakawa; Toshikatsu, Otani (1964). «Properties of Various Pure Irons: Study on pure iron I». Tetsu-to-Hagane. 50 (1): 42–47. doi:10.2355/tetsutohagane1955.50.1_42.
- ^ Raghavan, V. (2004). Materials Science and Engineering. PHI Learning Pvt. Ltd. p. 218. ISBN 81-203-2455-2.
- ^ Martin, John Wilson (2007). Concise encyclopedia of the structure of materials. Elsevier. p. 183. ISBN 978-0-08-045127-5.
- ^ a b Camp, James McIntyre; Francis, Charles Blaine (1920). The Making, Shaping and Treating of Steel. Pittsburgh: Carnegie Steel Company. pp. 173–74. ISBN 1-147-64423-3.
- ^ a b Smith, William F.; Hashemi, Javad (2006), Foundations of Materials Science and Engineering (4th ed.), McGraw-Hill, p. 431, ISBN 0-07-295358-6.
- ^ a b «Classification of Carbon and Low-Alloy Steels». Archived from the original on 2 January 2011. Retrieved 5 January 2008.
- ^ HSLA Steel, 15 November 2002, archived from the original on 30 December 2009, retrieved 11 October 2008.
- ^ Oberg, E.; et al. (1996), «Machinery’s Handbook», New York: Industrial Press (25th ed.), Industrial Press Inc: 440–42, Bibcode:1984msh..book…..R
- ^ Rokni, Sayed H.; Cossairt, J. Donald; Liu, James C. (January 2008). «Radiation Shielding at High-Energy Electron and Proton Accelerators» (PDF). Retrieved 6 August 2016.
- ^ a b c Greenwood & Earnshaw 1997, p. 1076.
- ^ Fürstner, Alois (2016). «Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes to Make This Base Metal a Multitasking Champion». ACS Central Science. 2 (11): 778–789. doi:10.1021/acscentsci.6b00272. PMC 5140022. PMID 27981231.
- ^ Bullock, R. Morris; et al. (2020). «Using nature’s blueprint to expand catalysis with Earth-abundant metals». Science. 369 (6505): eabc3183. doi:10.1126/science.abc3183. PMC 7875315. PMID 32792370.
- ^ Kolasinski, Kurt W. (2002). «Where are Heterogenous Reactions Important». Surface science: foundations of catalysis and nanoscience. John Wiley and Sons. pp. 15–16. ISBN 978-0-471-49244-3.
- ^ McKetta, John J. (1989). «Nitrobenzene and Nitrotoluene». Encyclopedia of Chemical Processing and Design: Volume 31 – Natural Gas Liquids and Natural Gasoline to Offshore Process Piping: High Performance Alloys. CRC Press. pp. 166–67. ISBN 978-0-8247-2481-8.
- ^ a b c Wildermuth, Egon; Stark, Hans; Friedrich, Gabriele; Ebenhöch, Franz Ludwig; Kühborth, Brigitte; Silver, Jack; Rituper, Rafael (2000). «Iron Compounds». Ullmann’s Encyclopedia of Industrial Chemistry. doi:10.1002/14356007.a14_591. ISBN 3-527-30673-0.
- ^ Stroud, Robert (1933). Diseases of Canaries. Canary Publishers Company. p. 203. ISBN 978-1-4465-4656-7.
- ^ World Health Organization (2021). World Health Organization model list of essential medicines: 22nd list (2021). Geneva: World Health Organization. hdl:10665/345533. WHO/MHP/HPS/EML/2021.02.
- ^ Dlouhy, Adrienne C.; Outten, Caryn E. (2013). Banci, Lucia (ed.). Metallomics and the Cell. Metal Ions in Life Sciences. Vol. 12. Springer. pp. 241–78. doi:10.1007/978-94-007-5561-1_8. ISBN 978-94-007-5560-4. PMC 3924584. PMID 23595675. electronic-book ISBN 978-94-007-5561-1
- ^
Yee, Gereon M.; Tolman, William B. (2015). Peter M.H. Kroneck; Martha E. Sosa Torres (eds.). Sustaining Life on Planet Earth: Metalloenzymes Mastering Dioxygen and Other Chewy Gases. Metal Ions in Life Sciences. Vol. 15. Springer. pp. 131–204. doi:10.1007/978-3-319-12415-5_5. PMID 25707468. - ^ a b c d e f g h i j k l m n o p Greenwood & Earnshaw 1997, pp. 1098–104.
- ^ Lippard, S.J.; Berg, J.M. (1994). Principles of Bioinorganic Chemistry. Mill Valley: University Science Books. ISBN 0-935702-73-3.
- ^ Kikuchi, G.; Yoshida, T.; Noguchi, M. (2005). «Heme oxygenase and heme degradation». Biochemical and Biophysical Research Communications. 338 (1): 558–67. doi:10.1016/j.bbrc.2005.08.020. PMID 16115609.
- ^
Uebe, René; Schüler, Dirk; «The Formation of Iron Biominerals «, pp 159-184 in «Metals, Microbes and Minerals: The Biogeochemical Side of Life» (2021) pp xiv + 341. Walter de Gruyter, Berlin. Editors Kroneck, Peter M.H. and Sosa Torres, Martha. DOI 10.1515/9783110589771-006 - ^ Neilands, J.B. (1995). «Siderophores: structure and function of microbial iron transport compounds». The Journal of Biological Chemistry. 270 (45): 26723–26. doi:10.1074/jbc.270.45.26723. PMID 7592901.
- ^ Neilands, J.B. (1981). «Microbial Iron Compounds». Annual Review of Biochemistry. 50 (1): 715–31. doi:10.1146/annurev.bi.50.070181.003435. PMID 6455965.
- ^ Boukhalfa, Hakim; Crumbliss, Alvin L. (2002). «Chemical aspects of siderophore mediated iron transport». BioMetals. 15 (4): 325–39. doi:10.1023/A:1020218608266. PMID 12405526. S2CID 19697776.
- ^ Nanami, M.; Ookawara, T.; Otaki, Y.; Ito, K.; Moriguchi, R.; Miyagawa, K.; Hasuike, Y.; Izumi, M.; Eguchi, H.; Suzuki, K.; Nakanishi, T. (2005). «Tumor necrosis factor-α-induced iron sequestration and oxidative stress in human endothelial cells». Arteriosclerosis, Thrombosis, and Vascular Biology. 25 (12): 2495–501. doi:10.1161/01.ATV.0000190610.63878.20. PMID 16224057.
- ^ Rouault, Tracey A. (2003). «How Mammals Acquire and Distribute Iron Needed for Oxygen-Based Metabolism». PLOS Biology. 1 (3): e9. doi:10.1371/journal.pbio.0000079. PMC 300689. PMID 14691550.
- ^ Boon EM, Downs A, Marcey D. «Proposed Mechanism of Catalase». Catalase: H2O2: H2O2 Oxidoreductase: Catalase Structural Tutorial. Retrieved 11 February 2007.
- ^ Boyington JC, Gaffney BJ, Amzel LM (1993). «The three-dimensional structure of an arachidonic acid 15-lipoxygenase». Science. 260 (5113): 1482–86. Bibcode:1993Sci…260.1482B. doi:10.1126/science.8502991. PMID 8502991.
- ^ Gray, N.K.; Hentze, M.W. (August 1994). «Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs». EMBO J. 13 (16): 3882–91. doi:10.1002/j.1460-2075.1994.tb06699.x. PMC 395301. PMID 8070415.
- ^ Gregory B. Vásquez; Xinhua Ji; Clara Fronticelli; Gary L. Gilliland (1998). «Human Carboxyhemoglobin at 2.2 Å Resolution: Structure and Solvent Comparisons of R-State, R2-State and T-State Hemoglobins». Acta Crystallogr. D. 54 (3): 355–66. doi:10.1107/S0907444997012250. PMID 9761903.
- ^ Sanderson, K (2017). «Mussels’ iron grip inspires strong and stretchy polymer». Chemical & Engineering News. American Chemical Society. 95 (44): 8. doi:10.1021/cen-09544-notw3. Retrieved 2 November 2017.
- ^ Food Standards Agency – Eat well, be well – Iron deficiency Archived 8 August 2006 at the Wayback Machine. Eatwell.gov.uk (5 March 2012). Retrieved on 27 June 2012.
- ^ Hoppe, M.; Hulthén, L.; Hallberg, L. (2005). «The relative bioavailability in humans of elemental iron powders for use in food fortification». European Journal of Nutrition. 45 (1): 37–44. doi:10.1007/s00394-005-0560-0. PMID 15864409. S2CID 42983904.
- ^ Pineda, O.; Ashmead, H. D. (2001). «Effectiveness of treatment of iron-deficiency anemia in infants and young children with ferrous bis-glycinate chelate». Nutrition. 17 (5): 381–4. doi:10.1016/S0899-9007(01)00519-6. PMID 11377130.
- ^ Ashmead, H. DeWayne (1989). Conversations on Chelation and Mineral Nutrition. Keats Publishing. ISBN 0-87983-501-X.
- ^ Institute of Medicine (US) Panel on Micronutrients (2001). «Iron» (PDF). Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Iron. National Academy Press. pp. 290–393. ISBN 0-309-07279-4. PMID 25057538. Archived from the original (PDF) on 9 September 2017. Retrieved 9 March 2017.
- ^ «Overview on Dietary Reference Values for the EU population as derived by the EFSA Panel on Dietetic Products, Nutrition and Allergies» (PDF). European Food Safety Authority. 2017.
- ^ «Tolerable Upper Intake Levels For Vitamins And Minerals» (PDF). European Food Safety Authority. 2006.
- ^ «Iron Deficiency Anemia». MediResource. Retrieved 17 December 2008.
- ^ Milman, N. (1996). «Serum ferritin in Danes: studies of iron status from infancy to old age, during blood donation and pregnancy». International Journal of Hematology. 63 (2): 103–35. doi:10.1016/0925-5710(95)00426-2. PMID 8867722.
- ^ «Federal Register May 27, 2016 Food Labeling: Revision of the Nutrition and Supplement Facts Labels. FR page 33982» (PDF).
- ^ «Daily Value Reference of the Dietary Supplement Label Database (DSLD)». Dietary Supplement Label Database (DSLD). Archived from the original on 7 April 2020. Retrieved 16 May 2020.
- ^ Centers for Disease Control and Prevention (2002). «Iron deficiency – United States, 1999–2000». MMWR. 51 (40): 897–99. PMID 12418542.
- ^ Hider, Robert C.; Kong, Xiaole (2013). «Chapter 8. Iron: Effect of Overload and Deficiency». In Astrid Sigel, Helmut Sigel and Roland K.O. Sigel (ed.). Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences. Vol. 13. Springer. pp. 229–94. doi:10.1007/978-94-007-7500-8_8. PMID 24470094.
- ^ Dlouhy, Adrienne C.; Outten, Caryn E. (2013). «Chapter 8.4 Iron Uptake, Trafficking and Storage». In Banci, Lucia (ed.). Metallomics and the Cell. Metal Ions in Life Sciences. Vol. 12. Springer. pp. 241–78. doi:10.1007/978-94-007-5561-1_8. ISBN 978-94-007-5560-4. PMC 3924584. PMID 23595675. electronic-book ISBN 978-94-007-5561-1
- ^ CDC Centers for Disease Control and Prevention (3 April 1998). «Recommendations to Prevent and Control Iron Deficiency in the United States». Morbidity and Mortality Weekly Report. 47 (RR3): 1. Retrieved 12 August 2014.
- ^ Centers for Disease Control and Prevention. «Iron and Iron Deficiency». Retrieved 12 August 2014.
- ^ Ramzi S. Cotran; Vinay Kumar; Tucker Collins; Stanley Leonard Robbins (1999). Robbins pathologic basis of disease. Saunders. ISBN 978-0-7216-7335-6. Retrieved 27 June 2012.
- ^ Ganz T (August 2003). «Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation». Blood. 102 (3): 783–8. doi:10.1182/blood-2003-03-0672. PMID 12663437. S2CID 28909635.
- ^ Durupt, S.; Durieu, I.; Nové-Josserand, R.; Bencharif, L.; Rousset, H.; Vital Durand, D. (2000). «Hereditary hemochromatosis». Rev Méd Interne. 21 (11): 961–71. doi:10.1016/S0248-8663(00)00252-6. PMID 11109593.
- ^ a b Cheney, K.; Gumbiner, C.; Benson, B.; Tenenbein, M. (1995). «Survival after a severe iron poisoning treated with intermittent infusions of deferoxamine». J Toxicol Clin Toxicol. 33 (1): 61–66. doi:10.3109/15563659509020217. PMID 7837315.
- ^ a b «Toxicity, Iron». Medscape. Retrieved 23 May 2010.
- ^ Dietary Reference Intakes (DRIs): Recommended Intakes for Individuals (PDF), Food and Nutrition Board, Institute of Medicine, National Academies, 2004, archived from the original (PDF) on 14 March 2013, retrieved 9 June 2009
- ^ Tenenbein, M. (1996). «Benefits of parenteral deferoxamine for acute iron poisoning». J Toxicol Clin Toxicol. 34 (5): 485–89. doi:10.3109/15563659609028005. PMID 8800185.
- ^ Wu H, Wu T, Xu X, Wang J, Wang J (May 2011). «Iron toxicity in mice with collagenase-induced intracerebral hemorrhage». J Cereb Blood Flow Metab. 31 (5): 1243–50. doi:10.1038/jcbfm.2010.209. PMC 3099628. PMID 21102602.
- ^ Robberecht, Harry; et al. (2020). «Magnesium, Iron, Zinc, Copper and Selenium Status in Attention-Deficit/Hyperactivity Disorder (ADHD)». Molecules. 25 (19): 4440. doi:10.3390/molecules25194440. PMC 7583976. PMID 32992575.
- ^ Soto-Insuga, V; et al. (2013). «[Role of iron in the treatment of attention deficit-hyperactivity disorder]». An Pediatr (Barc). 79 (4): 230–235. doi:10.1016/j.anpedi.2013.02.008. PMID 23582950.
- ^ Parisi, Pasquale; Villa, Maria Pia; Donfrancesco, Renato; Miano, Silvia; Paolino, Maria Chiara; Cortese, Samuele (August 2012). «Could treatment of iron deficiency both improve ADHD and reduce cardiovascular risk during treatment with ADHD drugs?». Medical Hypotheses. 79 (2): 246–249. doi:10.1016/j.mehy.2012.04.049. PMID 22632845.
- ^ Donfrancesco, Renato; Parisi, Pasquale; Vanacore, Nicola; Martines, Francesca; Sargentini, Vittorio; Cortese, Samuele (May 2013). «Iron and ADHD: Time to Move Beyond Serum Ferritin Levels». Journal of Attention Disorders. 17 (4): 347–357. doi:10.1177/1087054711430712. ISSN 1087-0547. PMID 22290693. S2CID 22445593.
- ^ Thévenod, Frank (2018). «Chapter 15. Iron and Its Role in Cancer Defense: A Double-Edged Sword». In Sigel, Astrid; Sigel, Helmut; Freisinger, Eva; Sigel, Roland K. O. (eds.). Metallo-Drugs: Development and Action of Anticancer Agents. Metal Ions in Life Sciences. Vol. 18. Berlin: de Gruyter GmbH. pp. 437–67. doi:10.1515/9783110470734-021. PMID 29394034.
- ^ a b Beguin, Y; Aapro, M; Ludwig, H; Mizzen, L; Osterborg, A (2014). «Epidemiological and nonclinical studies investigating effects of iron in carcinogenesis—a critical review». Critical Reviews in Oncology/Hematology. 89 (1): 1–15. doi:10.1016/j.critrevonc.2013.10.008. PMID 24275533.
- ^ Morel, F.M.M., Hudson, R.J.M., & Price, N.M. (1991). Limitation of productivity by trace metals in the sea. Limnology and Oceanography, 36(8), 1742-1755. doi:10.4319/lo.1991.36.8.1742
- ^ Brezezinski, M.A., Baines, S.B., Balch, W.M., Beucher, C.P., Chai, F., Dugdale, R.C., Krause, J.W., Landry, M.R., Marchi, A., Measures, C.I., Nelson, D.M., Parker, A.E., Poulton, A.J., Selph, K.E., Strutton, P.G., Taylor, A.G., & Twining, B.S.(2011). Co-limitation of diatoms by iron and silicic acid in the equatorial Pacific. Deep-Sea Research Part II: Topical Studies in Oceanography, 58(3-4), 493-511. doi:10.1016/j.dsr2.2010.08.005
- ^ Field, E. K., Kato, S., Findlay, A. J., MacDonald, D. J., Chiu, B. K., Luther, G. W., & Chan, C. S. (2016). Planktonic marine iron oxidizers drive iron mineralization under low-oxygen conditions. Geobiology, 14(5), 499-508. doi:10.1111/gbi.12189
- ^ Wells, M.L., Price, N.M., & Bruland, K.W. (1995). Iron chemistry in seawater and its relationship to phytoplankton: a workshop report. Marine Chemistry, 48(2), 157-182. doi:10.1016/0304-4203(94)00055-I
- ^ Lannuzel, D., Vancoppenolle, M., van der Merwe, P., de Jong, J., Meiners, K.M., Grotti, M., Nishioska, J., & Schoemann. (2016). Iron in sea ice: Review and new insights. Elementa: Science of the Anthropocene, 4 000130.
doi:10.12952/journal.elementa.000130 - ^ Raiswell, R. 2011. Iron Transport from the Continents to the Open Ocean: The Aging–Rejuvenation Cycle. Elements, 7(2), 101–106. doi:10.2113/gselements.7.2.101
- ^ Tagliabue, A., Bopp, L., Aumont,O., & Arrigo, K.R. (2009). Influence of light and temperature on the marine iron cycle: From theoretical to global modeling. Global Biogeochemical Cycles, 23.
doi:10.1029/2008GB003214
Bibliography
- Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 978-0-08-037941-8.
- Weeks, Mary Elvira; Leichester, Henry M. (1968). «Elements known to the ancients». Discovery of the elements. Easton, PA: Journal of Chemical Education. pp. 29–40. ISBN 0-7661-3872-0. LCCN 68-15217.
Further reading
- H.R. Schubert, History of the British Iron and Steel Industry … to 1775 AD (Routledge, London, 1957)
- R.F. Tylecote, History of Metallurgy (Institute of Materials, London 1992).
- R.F. Tylecote, «Iron in the Industrial Revolution» in J. Day and R.F. Tylecote, The Industrial Revolution in Metals (Institute of Materials 1991), 200–60.
External links
![]()
Wikiquote has quotations related to Iron.
![]()
Look up iron in Wiktionary, the free dictionary.
![]()
Wikimedia Commons has media related to Iron.
![]()
- It’s Elemental – Iron
- Iron at The Periodic Table of Videos (University of Nottingham)
- Metallurgy for the non-Metallurgist
- Iron by J.B. Calvert
Железо в таблице менделеева занимает 26 место, в 4 периоде.
| Символ | Fe |
| Номер | 26 |
| Атомный вес | 55.8450000 |
| Латинское название | Ferrum |
| Русское название | Железо |
Как самостоятельно построить электронную конфигурацию? Ответ здесь
Электронная схема железа
Fe: 1s2 2s2 2p6 3s2 3p6 4s2 3d6
Короткая запись:
Fe: [Ar]4s2 3d6
Одинаковую электронную конфигурацию имеют
атом железа и
Mn-1, Co+1, Ni+2
Порядок заполнения оболочек атома железа (Fe) электронами:
1s → 2s → 2p → 3s → 3p → 4s → 3d → 4p → 5s → 4d →
5p → 6s → 4f → 5d → 6p → 7s → 5f → 6d → 7p.
На подуровне ‘s’ может находиться до 2 электронов, на ‘s’ — до 6, на
‘d’ — до 10 и на ‘f’ до 14
Железо имеет 26 электронов,
заполним электронные оболочки в описанном выше порядке:
2 электрона на 1s-подуровне
2 электрона на 2s-подуровне
6 электронов на 2p-подуровне
2 электрона на 3s-подуровне
6 электронов на 3p-подуровне
2 электрона на 4s-подуровне
6 электронов на 3d-подуровне
Степень окисления железа
Атомы железа в соединениях имеют степени окисления 6, 5, 4, 3, 2, 1, 0, -1, -2.
Степень окисления — это условный заряд атома в соединении: связь в молекуле
между атомами основана на разделении электронов, таким образом, если у атома виртуально увеличивается
заряд, то степень окисления отрицательная (электроны несут отрицательный заряд), если заряд уменьшается,
то степень окисления положительная.
Ионы железа
Валентность Fe
Атомы железа в соединениях проявляют валентность VI, V, IV, III, II, I.
Валентность железа характеризует способность атома Fe к образованию хмических связей.
Валентность следует из строения электронной оболочки атома, электроны, участвующие в образовании
химических соединений называются валентными электронами. Более обширное определение валентности это:
Число химических связей, которыми данный атом соединён с другими атомами
Валентность не имеет знака.
Квантовые числа Fe
Квантовые числа определяются последним электроном в конфигурации,
для атома Fe эти числа имеют значение N = 3, L = 2, Ml = 3, Ms = -½
Видео заполнения электронной конфигурации (gif):
Результат:
Энергия ионизации
Чем ближе электрон к центру атома — тем больше энергии необходимо, что бы его оторвать.
Энергия, затрачиваемая на отрыв электрона от атома называется энергией ионизации и обозначается Eo.
Если не указано иное, то энергия ионизации — это энергия отрыва первого электрона, также существуют энергии
ионизации для каждого последующего электрона.
Энергия ионизации Fe:
Eo = 763 кДж/моль
— Что такое ион читайте в статье.
Перейти к другим элементам таблицы менделеева
Где Fe в таблице менделеева?
Таблица Менделеева
Скачать таблицу менделеева в хорошем качестве
Химический элемент с атомным номером 26
Химический элемент с атомным номером 26
| | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Железо | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Внешний вид | блестящий металлик с сероватым оттенком | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Стандартный атомный вес A r, std (Fe) | 55,845 (2) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Железо в таблица Менделеева | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Атомный номер (Z) | 26 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Группа | группа 8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Период | период 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| d-блок | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Категория элемента | Переходный металл | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Электронная конфигурация | [Ar ] 3d 4s | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Электронов на оболочку | 2, 8, 14, 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Физические | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Фаза при STP | свойства твердого тела | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Точка плавления | 1811 K (1538 ° C, 2800 ° F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Точка кипения | 3134 K (2862 ° C, 5182 ° F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Плотность (около rt ) | 7,874 г / см | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| в жидком состоянии (при т.пл. ) | 6,98 г / см | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Теплота плавления | 13,81 кДж / моль | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Теплота испарения | 340 кДж / моль | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Молярная теплоемкость | 25,10 Дж / (моль · К) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Давление пара
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Атомные свойства | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Степени окисления | −4, −2, −1, 0, +1, +2, +3, +4, +5, +6, +7 (амф. Отерический оксид) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Электроотрицательность | Шкала Полинга: 1,83 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Энергии ионизации |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Атомный радиус | эмпирический: 126 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ковалентный радиус | Низкое вращение: 132 ± 3 мкм. Высокое вращение: 152 ± 6 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Другие | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Естественное происхождение | изначальное | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Кристаллическая структура | объ емно-центрированная кубическая (bcc)  . a = 286,65 pm . a = 286,65 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Кристаллическая структура | гранецентрированный куб (ГЦК)  . между 1185–1667 К; a = 364,680 пм . между 1185–1667 К; a = 364,680 пм | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Скорость звука тонкий стержень | 5120 м / с (при rt ) (электролитический) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Тепловое расширение | 11,8 мкм / (м · К) (при 25 ° C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Теплопроводность | 80,4 Вт / (м · К) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Удельное электрическое сопротивление | 96,1 нОм · м (при 20 ° C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| точка Кюри | 1043 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Магнитное упорядочение | ферромагнетик | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Модуль Юнга | 211 ГПа | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Модуль сдвига | 82 ГПа | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Объемный модуль | 170 ГПа | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Коэффициент Пуассона | 0,29 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Твердость по Моосу | 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Твердость по Виккерсу | 608 МПа | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Твердость по Бринеллю | 200–1180 МПа | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Номер CAS | 7439-89 -6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| История | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Открытие | до 5000 г. до н.э. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Основные изотопы железа | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ссылки |
Железо () — это химический элемент с символом Fe(от Latin : ferrum ) и атомный номер 26. Это металл , который принадлежит к первой переходной серии и группе 8 периодической таблицы. Это по массе самый распространенный элемент на Земле, прямо перед кислородом (32,1% и 30,1% соответственно), формируя большую часть внешний и внутренний сердечник. Это четвертый по частям элемент в земной коре.
В своем металлическом состоянии железо редко встречается в земной коре, ограничиваясь в отложении основными метеоритами. Железные руды, напротив, являются одними из самых распространенных в земном коре, хотя для извлечения из них пригодного для использования металла требуются печи или печи, способные нагреваться до 1500 ° C (2730 ° F) или выше, примерно на 500 ° C (900 ° F) выше, чем требуется для плавления меди. Люди начали осваивать этот процесс в Евразии только около 2000 г. до н.э., и использование железных инструментов и оружия начало вытеснять медные сплавы в некоторых регионах только около 1200 г. до н. э. Это событие считается переходом от бронзового века к железному веку. В современном мире сплавы железа, такие как сталь, нержавеющая сталь, чугун и специальные стали, являются На сегодняшний день это наиболее распространенные промышленные металлы из-за их механических свойств и низкой стоимости.
Безупречные и гладкие поверхности из чистого железа зеркально-серебристо-серые. Однако железо легко реагирует с кислородом и водой с образованием коричневого или черного гидратированного оксидов железа, широко известных как ржавчина. В отличие от оксидов некоторых других металлов, которые образуют пассивирующие слои , ржавчина занимает больше объема, металл чем, и отслаивается, подвергается воздействию свежей поверхности коррозии.
Тело взрослого человека содержит около 4 граммов (0,005% веса тела) железа, в основном в виде гемоглобина и миоглобина. Эти два белка играют важную роль в метаболизме позвоночных, соответственно транспорте кислорода кровью и хранении кислорода в мышцы. Для поддержания необходимых уровней метаболизм железа у человека требует минимального количества железа в рационе. Железо также является металлом в активном центре многих важных redox ферментов, связанных с клеточным дыханием и окислением и восстановлением у растений и животных..
С химической точки зрения наиболее распространенными степенями окисления железа являются железо (II) и железо (III). Железо обладает способностью других переходных металлов, включая другие элементы группы 8, рутений и осмий. Железо образует соединения в широком диапазоне степеней окисления , от -2 до +7. Железо также образует множество координационных соединений ; некоторые из них, такие как ферроцен, ферриоксалат и берлинская лазурь, имеют существенное промышленное, медицинское или исследовательское применение.
Содержание
- 1 Характеристики
- 1.1 Аллотропы
- 1.2 Точки плавления и кипения
- 1.3 Магнитные свойства
- 1.4 Изотопы
- 2 Происхождение и встречаемость в природе
- 2.1 Космогенез
- 2.2 Металлическое железо
- 2.3 Минералы мантии
- 2.4 Земная кора
- 3 Химия и соединения
- 3.1 Бинарные соединения
- 3.1.1 Оксиды и гидроксиды
- 3.1.2 Галогениды
- 3.2 Химический состав раствор
- 3.3 Координационные соединения
- 3.4 Металлоорганические соединения
- 3.5 Промышленное использование
- 3.1 Бинарные соединения
- 4 Этимология
- 5 История
- 5.1 Развитие металлургии железа
- 5.1.1 Метеоритное железо
- 5.1.2 Кованое железо
- 5.1.3 Чугун
- 5.1.4 Сталь
- 5.2 Основы современной химии
- 5.1 Развитие металлургии железа
- 6 Символическая роль
- 7 Производство металлического железа
- 7.1 Лабораторные направления
- 7.2 Основные промышленный путь
- 7.2.1 Доменная обработка
- 7.2.2 Сталеплавильное производство
- 7.3 Прямое восстановлен ие железа
- 7.4 Термитный процесс
- 8 Применения
- 8.1 В качестве конструкционного материала
- 8.1.1 Механизм химические свойства
- 8.1.2 Типы сталей и сплавов
- 8.2 Соединения железа
- 8.1 В качестве конструкционного материала
- 9 Биологическая и патологическая роль
- 9.1 Биохимия
- 9.2 Питание
- 9.2.1 Диета
- 9.2 Рекомендации по питанию
- 9.3 Дефицит
- 9.4 Избыточность
- 9.5 Рак
- 10 См.. Также
- 11 Ссылки
- 12 Библиография
- 13 Дополнительная литература
- 14 Внешние ссылки
Характеристики
Аллотропы
Зависимость молярного объема от давления для α-железа при комнатной температуре
Известно, по крайней мере, четыре аллотропа железа (разное расположение элементов в твердом теле), условно обозначаемые α, γ, δ и ε.
Низкое давление Фазовая диаграмма чистого железа
Первые три формы наблюдаются при обычных давлениях. Когда расплавленное железо охлаждается выше точки замерзания 1538 ° C, оно кристаллизуется в свой δ-аллотроп, который имеет объемно-центрированную кубическую (bcc) кристаллическую структуру. По мере охлаждения до 1394 ° C он превращается в аллотроп γ-железа, кристаллическую структуру кубической гранецентрированной (ГЦК) или аустенит. При температуре 912 ° C и ниже кристаллическая структура снова становится аллотропом α-железа с ОЦК.
Физические свойства железа при очень высоких давлениях и температурах также широко изучались, поскольку они имеют отношение к теориям о ядре Земли и других планет. Выше приблизительно 10 ГПа и температура в несколько сотен градусов Кельвина или ниже α-железо превращается в другую гексагональную плотноупакованную (ГПУ) структуру, которая также известна как ε-железо. Γ-фаза при более высокой температуре также превращается в ε-железо, но при более высоком давлении.
Существуют противоречивые экспериментальные приборы существования стабильной β-фазы при давлении выше 50 ГПа и температурах не менее 1500 К. Предполагается, что она имеет орторомбическую или двойную ГПУ структуру. (Что сбивает с толку, термин «β-железо» иногда также используется для обозначения α-железа выше точки Кюри, когда оно изменяется с ферромагнитного на парамагнитное, если даже его кристаллическая структура не изменилась.)
внутреннееро объекта Земля обычно юридически ответственным из ядра железо никель со структурой ε (или β).
Точки плавления и кипения
Точки плавления и кипения, наряду с его энтальпией распыления, ниже, чем у более ранних 3d-элементов из скандия. От до хром, демонстрируя уменьшение вклада 3d-электронов в металлических связях, поскольку они все больше и больше притягиваются ядром к инертному ядру; однако они выше, чем значения для предыдущего элемента марганец, потому что этот элемент имеет наполовину заполненную трехмерную подоболочку и, следовательно, его d-электроны нелегко делокализовать. Та же самая тенденция проявляется для рутения, но не для осмия.
Температура плавления железа экспериментально хорошо определена для давлений менее 50 ГПа. Для более высоких давлений опубликованные данные (по состоянию на 2007 год) все еще являются на десятки гигапаскалей и более тысячи кельвинов.
Магнитные свойства
Кривые намагничивания 9 ферромагнитных материалов, показывающие насыщение. 1. Листовая сталь, 2. Кремниевая сталь, 3. Литая сталь, 4. Вольфрамовая сталь, 5. Магнитная сталь, 6. Чугун, 7. Никель, 8. Кобальт, 9. Магнетит
Ниже его Кюри точка 770 ° C, α-железо изменяется с парамагнитного на ферромагнитного : спины двух неспаренных электронов в каждом атоме обычно совпадают с вращает своих соседей, создавая общее магнитное поле. Это происходит потому, что орбитали эти двух электронов (d z и d x — y) не указывают на соседние атомы в решетке и, следовательно, не участвуют в металлических связях.
В отсутствие внешнего магнитного поля атомы спонтанно разделяются на магнитные домены диаметром около 10 микрометров, так что атомы в каждом домене имеют параллельные спины, но некоторые домены есть другие ориентации. Таким образом, макроскопический железа будет иметь почти нулевое общее магнитное поле.
Приложение внешнего магнитного поля заставляет домены, намагниченные в одном и том же общем направлении, расти за счет соседних, которые указывают в других направлениях, усиливая внешнее поле. Этот эффект используется в устройствах, через которые необходимо направлять магнитные поля, такие как электрические трансформаторы, головки магнитной записи и электродвигатели. Примеси, дефекты решетки или границы зерен и частиц могут «закрепить» домены в новых положениях, так что эффект сохраняется даже после удаления внешнего поля, превращая железный объект в (постоянный) магнит.
Подобное поведение демонстрируют некоторые соединения железа, такие как ферриты и минерал магнетит, кристаллическая форма смешанного оксида железа (II, III) Fe. 3O.. 4 (хотя механизм атомного масштаба, ферримагнетизм, несколько отличается). Куски магнетита с естественной намагниченностью (магниты ) послужили первыми компасами для навигации. Частицы магнетита широко использовались в магнитных носителях записи, такие как основные запоминающие устройства, магнитные ленты, дискеты и диски, пока они не были заменены материалы на основе кобальта.
Изотопы
Железо содержит четыре стабильных изотопа : Fe (5,845% природного железа), Fe (91,754%), Fe (2,119%).) и Fe (0,282%). Также создано 20-30 искусственных изотопов. Из этих стабильных изотопов только Fe имеет ядерный спин (- ⁄ 2). Нуклид Fe теоретически может подвергаться двойному захвату электронов в Cr, но этот процесс никогда не наблюдался, и был установлен только нижний предел периода полураспада 3,1 × 10 лет.
Fe представляет собой потухший радионуклид с длительным периодом полураспада (2,6 миллиона лет). Его нет на Земле, но конечным продуктом его распада является его внучка, стабильный нуклид Ni. Большая часть прошлых работ по изотопному составу железа была сосредоточена на нуклеосинтезе Fe посредством изучения метеоритов и образования руд. За последнее десятилетие достижения в области масс-спектрометрии позволяли обнаруживать и количественно определять небольшие естественные вариации наций стабильных изотопов железа. Большая часть этой работы проводится сообществами Земля и планетологами, появляются приложения к биологическим и промышленным системам.
В фазах метеоритов Семаркона и Червоны, корреляция между концентрацией Ni, внучкой Fe и обилием стабильных изотопов железа обеспечила доказательства существования Fe во время образования Солнечной системы. Возможно, энергия, выделяемая при распаде Fe, наряду с энергией, выделяющейся Al, способствовала переплавлению и дифференциации астероидов после их образования 4,6 миллиарда лет назад.. Обилие Ni, присутствующего во внеземном материале, может пролить свет на происхождение и раннюю историю Солнечной системы.
. Самый распространенный изотоп железа Fe представляет собой наиболее распространенный интерес для ученых-ядерщиков, потому что он представляет собой наиболее частую конечную точку нуклеосинтеза. Ni (14 альфа-частицы ) легко образуется из более легких ядер в альфа-процессе в ядерных реакциях в сверхновых (см. процесс горения кремния ), это конечная точка цепочек слияния внутри массивных звезд, поскольку добавление еще одной альфа-частицы, приводящей к образованию Zn, требует гораздо больше энергии. Этот Ni, период полураспада которого составляет около 6 дней, создается в больших количествах в этой звезде, но вскоре распадается в результате двух последовательных излучений позронов в продуктах распадановой в газовом облаке остатка сверхновой, сначала до радиоактивного Co, а затем до стабильного Fe. Таким образом, самый распространенный в ядре красных гигантов и наиболее распространенным металлом в железных метеоритах и в плотных металлических железных метеоритах и в плотных металлических железных метеоритах и в плотных металлических железных планетах, например Земля. Он также очень распространен во Вселенной по сравнению с другими стабильными металлами примерно с таким же атомным весом. Железо является шестым по содержанию . во Вселенной и наиболее распространенным огнеупорным элементом
Хотя дальнейшее небольшое увеличение энергии может быть извлеченный путем получения Ni, который имеет немного более высокую энергию связи, чем Fe, условия в звездах не подходят для этого процесса. Производство элементов в сверхновых звездах и их распространение на Земле в степени предпочтительнее для железа, чем для никеля, и в любом случае Fe по-прежнему имеет меньшую массу на нуклон, чем Ni, из-за более высокой степени более легких протонов. Следовательно, элементы тяжелее железа требуют сверхновой для их образования, включая быстрый захват нейтронов стартовыми ядрами Fe.
В далеком будущем Вселенной, предполагая, что распад протона не происходит, холодный синтез, происходящий посредством квантового туннелирования, заставил бы легкие ядра в обычном веществе сливаться с ядрами Fe. Деление и испускание альфа-частиц привело бы к распаду тяжелых ядер в железо, превращая все объекты звездной массы в холодные сферы из чистого железа.
Происхождение и появление в природе
Космогенез
Изобилие железа на каменистых планетах, таких как Земля, связано с его обильным производством в результате синтеза в больших звездах, где оно находится последний элемент, который будет произведен с высвобождением энергии перед сильным взрывом типа Ia сверхновой, который разбросает железо в космос. Другие типы сверхновых не влияют существенно на содержание железа (см. сверхновая звезда с коллапсом ядра ).
Металлическое железо
Полированный и химически протравленный кусок железного метеорита, который, как полагают, по составу похож на металлическое ядро Земли., демонстрирующие отдельные кристаллы железо-никелевого сплава (узор Видманштаттена ).
Металлическое или самородное железо редко встречается на поверхности Земли, потому что оно имеет тенденцию к окислению. Однако и то, и другое на Земле внутреннее и внешнее ядро , на долю которых приходится 35% массы всей Земли, как полагают, в основном состоят из сплава железа, возможно с никелем. Считается, что токи в жидком внешнем ядре являются источником магнитного поля Земли. Другие планеты земной группы (Меркурий, Венера, и Марс ), а также Луна, как полагают, имеют металлическое ядро, состоящее в основно м из железа. Также полагают, что астероиды M-типа быть частично или большей частью из сплава металлического железа.
Редкие железные метеориты — основная форма природного металлического железа на поверхности Земли. Изделия из холоднодеформированного метеоритного железа были найдены на различных археологических раскопках, относящихся к тем временам, когда выплавка железа еще не была развита; и инуиты в Гренландии, как сообщается, использовали железо из метеорита Кейп-Йорк для изготовления инструментов и охотничьего оружия. Примерно 1 из 20 метеоритов состоит из уникальных железоникелевых минералов тенита (35–80% железа) и камасита (90–95% железа). Самородное железо также редко встречается в базальтах, которые образовались из магм, которые вступили в контакт с богатыми углеродом осадочными породами, которые уменьшили летучесть кислорода в достаточной степени для кристаллизации железа. Он известен как теллурическое железо и описано в нескольких местах, таких как остров Диско в Западной Гренландии, Якутия в России и Бюль в Германии.
Минералы мантии
Ферропериклаз (Mg, Fe) O, твердый раствор периклаза (MgO) и вюстита (FeO), составляет около 20 % от объема нижней мантии Земли, что делает ее второй по распространенности минеральной фазой в этой области после силикатного перовскита (Mg, Fe) SiO 3 ; он также является основным носителем железа в нижней мантии. В нижней части переходной зоны мантии происходит реакция γ- (Mg, Fe) 2 [SiO 4 ] ↔ (Mg, Fe) [ SiO 3 ] + (Mg, Fe) O превращает γ-оливин в смесь силикатного перовскита и ферропериклаза и наоборот. В литературе эту минеральную фазу нижней мантии также часто называют магнезиовюститом. Силикатный перовскит может образовывать до 93% нижней мантии, а магниево-железо образует (Mg, Fe) SiO 3, считается самым распространенным минералом на Земле, составляющим 38% от ее объема.
Земная кора
Охровый путь в Руссильоне.
Хотя железо является самым распространенным элементом на Земле, большая часть этого железа сосредоточена во внутреннем и внешнем ядрах. Доля железа, содержащаяся в земной коре, составляет всего около 5% от общей массы коры и, таким образом, является только четвертым по распространенности элементом в этом слое (после кислорода, кремний и алюминий ).
Большая часть железа в коре объединяется с различными другими элементами, образуя множество минералов железа. Важным классом является железо оксидные минералы, такие как гематит (Fe 2O3), магнетит (Fe 3O4) и сидерит (FeCO 3), которые являются основными железными рудами. Многие изверженные породы также содержат сульфидные минералы пирротин и пентландит. 289>при выветривании железо имеет тенденцию выщелачиваться из сульфидных отложений в виде сульфата и из силикатных отложений в виде бикарбоната. Оба они окисляются в водном растворе и выпадают в осадок даже при слегка повышенном pH в виде оксида железа (III).
Пол осчатая формация железа в Миннесоте.
Крупные месторождения железа полосчатые железные образования, тип породы, состоящий из повторяющихся тонких слоев оксидов железа, чередующихся с полосами бедных железом сланцев и сланцев. Полосчатые образования железа были заложены в период между 3700 миллионами лет назад и 1800 миллионами лет назад.
Материалы, содержащие тонкоизмельченные оксиды или оксиды железа (III) -гидроксиды, такие как охра, использовались в качестве желтых, красных и коричневых пигментов с доисторических времен. Они также влияют на цвет различных горных пород и глин, включая целые геологические образования, такие как Painted Hills в Oregon и Buntsandstein («цветной песчаник», англ. Bunter ). Через Eisensandstein (юрский «железный песчаник», например, из Донздорфа в Германии) и банный камень в Великобритании, соединения железа ответственны за желтоватый цвет много исторических зданий и скульптур. Известный красный цвет поверхности Марса происходит из-за богатого оксидом железа реголита.
. Значительное количество железа содержится в минерале сульфида железа пирите (FeS 2), но из него трудно извлечь железо, и поэтому он не используется. Фактически, железо настолько распространено, что производство обычно сосредоточено только на рудах с очень большим его содержанием.
Согласно отчету «Запасы металла в обществе» Международной группы ресурсов , глобальные запасы железа, используемого в обществе, составляют 2200 кг на душу населения. В этом отношении более развитые страны отличаются от менее развитых стран (7000–14000 против 2000 кг на душу населения).
Химия и соединения
| Окисление. состояние | Типичное соединение |
|---|---|
| -2 (d) | Тетракарбонилферрат динатрия (реагент Коллмана) |
| -1 (d) | Fe. 2(CO). 8 |
| 0 (d) | Пентакарбонил железа |
| 1 ( d) | Циклопентадиенилидикарбонилдимер железа («Fp 2 ») |
| 2 (d) | Сульфат железа, ферроцен |
| 3 (d) | Хлорид железа, тетрафторборат ферроцения |
| 4 (d) | Fe (диарс). 2Cl. 2 |
| 5 (d) | FeO. 4 |
| 6 ( d) | Феррат калия |
| 7 (d) | [FeO 4 ] (матричная изоляция, 4K) |
Железо демонстрирует характерные химические свойства переходные металлы, а именно способность образовывать различные степени окисления, различающиеся ступенями одной, и очень большой координационный и металлоорганический химический состав: действительно, именно открытие соединения железа, ферроцена, стало новым люционализировал последнее поле в 1950-х годах. Железо иногда считают прототипом всего блока переходных металлов из-за его большого количества и огромной роли, которую оно сыграло в техническом прогрессе человечества. Его 26 электронов расположены в конфигурации [Ar] 3d4s, из которых 3d- и 4s-электроны относительно близки по энергии, и, таким образом, он может потерять переменное количество электронов, и нет четкой точки, где дальше ионизация становится невыгодной.
Железо образует соединения в основном со степенями окисления +2 (железо (II), «двухвалентное») и +3 (железо (III), «железо»). Железо также присутствует в более высоких степенях окисления, например. фиолетовый феррат калия (K2FeO 4), который содержит железо в степени окисления +6. Хотя заявлен оксид железа (VIII) (FeO 4), отчет не может быть воспроизведен, и такой вид (по крайней мере, с железом в его степени окисления +8) оказался маловероятным с вычислительной точки зрения. Однако одна форма анионного соединения [FeO 4 ] с железом в степени окисления +7 вместе с (V) -пероксоизомером железа была обнаружена с помощью инфракрасной спектроскопии при 4 К после совместной конденсации лазерного излучения. удаляли атомы Fe смесью O 2 / Ar. Железо (IV) является обычным промежуточным продуктом во многих реакциях биохимического окисления. Многочисленные железоорганические соединения содержат формальные степени окисления +1, 0, -1 или даже -2. Степени окисления и другие связывающие свойства часто оценивают с использованием метода мессбауэровской спектроскопии. Многие соединения со смешанной валентностью содержат центры железа (II) и железа (III), такие как магнетит и берлинская лазурь (Fe 4 (Fe [CN] 6)3). Последний используется как традиционный «синий» в чертежах.
. Железо — первый из переходных металлов, который не может достичь своей групповой степени окисления +8, хотя и тяжелее конгенеры рутения и осмия могут, при этом рутений испытывает большие трудности, чем осмий. Рутений проявляет водно-катионный химический состав в его низких степенях окисления, аналогичный таковому у железа, но осмий нет, предпочитая высокие степени окисления, в которых он образует анионные комплексы. В половине серии переходов 3d вертикальное сходство вниз по группам соперничает с горизонтальным сходством железа с его соседями кобальтом и никелем в периодической таблице, которые также являются ферромагнитными на при комнатной температуре и имеют схожий химический состав. По этой пр ичине железо, кобальт и никель иногда группируются вместе как th e триада железа.
В отличие от многих других металлов, железо не образует амальгам с ртутью. В результате ртуть продается в стандартных 76-фунтовых колбах (34 кг), сделанных из железа.
Железо на сегодняшний день является наиболее реактивным элементом в своей группе; он пирофорен при мелком измельчении и легко растворяется в разбавленных кислотах, давая Fe. Однако он не реагирует с концентрированной азотной кислотой и другими окисляющими кислотами из-за образования непроницаемого оксидного слоя, который, тем не менее, может реагировать с соляной кислотой.
Бинарными соединениями
Оксиды и гидроксиды
Оксид двухвалентного железа или железа (II), FeO. Оксид трехвалентного железа или железа (III) Fe. 2O. 3. Железо или оксид железа (II, III) Fe. 3O. 4.
Формы железа различные оксидные и гидроксидные соединения ; наиболее распространенными являются оксид железа (II, III) (Fe 3O4) и оксид железа (III) (Fe 2O3). Оксид железа (II) также существует, хотя он нестабилен при комнатной температуре. Несмотря на их названия, на самом деле все они нестехиометрические соединения, состав которых может варьироваться. Эти оксиды являются основными рудами для производства железа (см. блюмерное производство и доменная печь). Они также используются в производстве ферритов, полезных магнитных запоминающих устройств в компьютерах и пигментов. Самый известный сульфид — железный пирит (FeS 2), также известный как золото дураков из-за его золотистого блеска. Это не соединение железа (IV), а фактически представляет собой полисульфид железа (II) , содержащий ионы Fe и S. 2в искаженной структуре хлорида натрия.
Pourbaix диаграмма железа
Галогениды
Хлорид гидратированного железа (III) (хлорид железа)
Бинарные галогениды двухвалентного и трехвалентного железа хорошо известны. Галогениды двухвалентного железа обычно образуются при обработке металлического железа соответствующей галогенводородной кислотой с получением соответствующих гидратированных солей.
- Fe + 2 HX → FeX 2 + H 2 (X = F, Cl, Br, I)
Железо реагирует с фтором, хлором и бромом с образованием соответствующих галогенидов железа, из которых хлорид железа является наиболее распространенным.
- 2 Fe + 3 X 2 → 2 FeX 3 (X = F, Cl, Br)
Иодид железа является исключением, поскольку он термодинамически нестабилен из-за окислительной способности Fe и высокая восстанавливающая способность I:
- 2 I + 2 Fe → I 2 + 2 Fe (E = +0,23 В)
Иодид железа, твердое вещество черного цвета, нестабилен в обычных условиях, но может быть получен реакцией пентакарбонила железа с йодом и монооксидом углерода в присутствии гексана и светом при температуре -20 ° C, без кислорода и воды.
Химический состав раствора
Сравнение цветов растворов феррата (слева) и permanganate (right)
The standard reduction potentials in acidic aqueous solution for some common iron ions are given below:
| Fe + 2 e | ⇌ Fe | E = −0.447 V |
| Fe + 3 e | ⇌ Fe | E = −0.037 V |
| FeO. 4+ 8 H + 3 e | ⇌ Fe + 4 H2O | E = +2.20 V |
The red-purple tetrahedral ferrate (VI) anion is such a strong oxidizing agent that it oxidizes nitrogen and ammonia at room temperature, and even water itself in acidic or neutral solutions:
- 4 FeO. 4+ 10 H. 2O → 4 Fe. + 20 OH. + 3 O2
The Fe ion has a large simple cationic chemistry, although the pale-violet hexaquo ion [Fe(H2O)6] is very readily hydrolyzed when pH increases above 0 as follows:
| [Fe(H2O)6] | ⇌ [Fe(H2O)5(OH)] + H | K = 10 mol dm |
| [Fe(H2O)5(OH)] | ⇌ [Fe(H2O)4(OH)2] + H | K = 10 mol dm |
| 2 [Fe(H2O)6] | ⇌ [Fe(H. 2O). 4(OH)]. 2+ 2 H + 2 H2O | K = 10 mol dm |
Blue-green iron(II) sulf ate heptahydrate
As pH rises above 0 the above yellow hydrolyzed species form and as it rises above 2–3, reddish-brown hydrous iron(III) oxide precipitates out of solution. Although Fe has an d configuration, its absorption spectrum is not like that of Mn with its weak, spin-forbidden d–d bands, because Fe has higher positive charge and is more polarizing, lowering the energy of its ligand-to-metal charge transfer absorptions. Thus, all the above complexes are rather strongly colored, with the single exception of the hexaquo ion – and even that has a spectrum dominated by charge transfer in the near ultraviolet region. On the other hand, the pale green iron(II) hexaquo ion [Fe(H2O)6] does not undergo appreciable hydrolysis. Carbon dioxide is not evolved when carbonate anions are added, which instead results in white iron(II) carbonate being precipitated out. In excess carbon dioxide this forms the slightly soluble bicarbonate, which occurs commonly in groundwater, but it oxidises quickly in air to form оксид железа (III), который составляет коричневые отложения, присутствующие в значительном количестве потоков.
Координационные соединения
Из-за своей электронной структуры железо имеет очень большую координационная и металлоорганическая химия.
Два энантиоморфа иона ферриоксалата
Известно много координационных соединений железа. Типичным шестикоординированным анионом является гексахлорферрат (III), [FeCl 6 ], обнаруженный в смешанной соли тетракис (метиламмоний) гексахлорферрат (III) хлорид. Комплексы с множественными бидентатными лигандами имеют геометрические изомеры. Например, комплекс транс- хлоргидридобис (бис-1,2- (дифенилфосфино) этан) железа (II) используется в качестве исходного материала для соединений с фрагментом Fe (dppe )2. Ферриоксалат-ион с тремя оксалатными лигандами (показан справа) демонстрирует спиральную хиральность с двумя несовместимыми геометриями, обозначенными Λ (лямбда) для оси левого винта. и Δ (дельта) для оси правого винта в соответствии с соглашениями IUPAC. Ферриоксалат калия используется в химической актинометрии и вместе с его натриевой солью подвергается фотовосстановлению, применяемому в старых фотографических процессах. дигидрат оксалата железа (II) имеет полимерную структуру с копланарной структурой. ионы оксалата, соединяющие центры железа с кристаллизационной водой, образующие верхушки каждого октаэдра, как показано ниже.
Кристаллическая структура дигидрата оксалата железа (II), показывающая железо (серый), кислород (красный), углерод (черный) k) и атомы водорода (белые). Кроваво-красный положительный тиоцианатный тест для железа (III)
Комплексы железа (III) очень похожи на комплексы хрома (III) с исключение предпочтения железа (III) к O-донору вместо лигандов N-донора. Последние имеют тенденцию быть более нестабильными, чем комплексы железа (II), и часто диссоциируют в воде. Многие комплексы Fe – O имеют интенсивный цвет и используются в качестве тестов для определения фенолов или енолов. Например, в тесте с хлоридом железа, используемом для определения присутствия фенолов, хлорид железа (III) реагирует с фенолом с образованием темно-фиолетового комплекса:
- 3 ArOH + FeCl 3 → Fe (OAr) 3 + 3 HCl (Ar = арил )
Среди галогенидных и псевдогалогенидных комплексов фторокомплексы железа (III) наиболее стабильными, при этом бесцветный [FeF 5(H2O)] является наиболее стабильным в водном растворе. Хлоркомплексы менее стабильной и способствуют тетраэдрической стране, как в [FeCl 4 ]; [FeBr 4 ] и [FeI 4 ] легко восстанавливаются до железа (II). Тиоцианат — это обычный тест на присутствие железа (III), поскольку он образует кроваво-красный [Fe (SCN) (H 2O)5]). Как и марганец (II), большинство комплексов железа (III) являются высокоспиновыми, за исключением тех, у которых лиганды имеют высокое содержание в спектрохимической серии, например, цианид. Примером низкоспинового комплекса железа (III) является [Fe (CN) 6 ]. Цианидные лиганды могут быть легко отделены в [Fe (CN) 6 ], и, следовательно, этот комплекс ядовит, в отличие от комплекса железа (II) [Fe (CN) 6 ], обнаруженный в берлинской синей, которая не выделяется цианистый водород, за исключением случаев добавления разбавленных кислот. Железо демонстрирует большое разнообразие электронных спиновых состояний, включая все возможные значения спинового квантового числа для элемента d-блока от 0 (диамагнитный) до ⁄ 2 (5 неспаренных электронов). Это значение всегда составляет половину числа неспаренных электронов. Комплексы от нуля до двух неспаренных электроны считаются низкоспиновыми, а электроны с четырьмя или пятью считаются высокоспиновыми..
Комплексы железа (II) менее стабильны, чем комплексы железа (III), но предпочтение О-донорных лигандов выражено, например, [Fe (NH 3)6]] известен, а [Fe (NH 3)6] — нет.
Металлоорганические соединения
Пента-. карбонил железа.
Они имеют тенденцию к окислению до железа (III), но это можно смягчить низким pH и используемыми специфическими лигандами.>Химия органического железа — это исследование металлоорганических соединений железа, в которых атомы углерода связаны с атомом металла. Их много и они разнообразны, в том числе цианидные комплексы, карбонильные комплексы, сэндвич и полусэндвич-соединения.
![]() берлинская лазурь.
берлинская лазурь.
берлинская лазурь. синий или «ферроцианид железа», Fe 4 [Fe (CN) 6]3, представляет собой старый и хорошо известный комплекс цианида железа, широко используется в качестве пигмента и в некоторых других областях. но использовать в качестве простого химического теста для определения различия между водными растворами Fe и Fe, как они реагируют (соответственно) с феррицианидом калия и ферроцианидом калия с образованием берлинской синей. 663>
Еще одним старым примером железоорганического соединения является пентакарбонил железа, Fe (CO) 5, в котором нейтральный атом железа связан с атомами углерода молекул оксида углерода. Соединение можно использовать для получения порошка карбонильного железа, высокореакционной формы металлического железа. Термолиз пентакарбонила железа дает додекакарбонил трижелеза, Fe. 3 (CO). 12, кластер из трех атомов железа в его ядре. Реагент Колмана, тетракарбонилферрат динатрия, является полезным реагентом для органической химии; он содержит железо в степени окисления -2. Циклопентадиенилдикарбонилдимер железа содержит железо в редкой степени окисления +1.
Структурная формула ферроцена и порошкообразный образец.
Знаковым в этой области стало открытие в 1951 г. удивительно стабильного сэндвич-соединение ферроцен Fe (C. 5H. 5). 2, автор] и независимо друг от друга, чья удивительная молекулярная структура была определена только спустя год Woodward и Уилкинсон и Фишер. Ферроцен по-прежнему одним из наиболее важных инструментов и моделей в этом классе.
Металлоорганические соединения с железоцентрированной структурой используются в качестве . Комплекс Кнёлькера, например, представляет катализатор гидрирования с переносом для кетонов.
Промышленное использование
Соединения железа, производимые на основе в промышленности используются сульфат железа (II) (FeSO 4·7H2O ) и хлорид железа (III) (FeCl 3). доступные источники железа (II), но она более устойчива к окислению в воздухе, чем соль Мора ((NH 4)2Fe (SO 4)2· 6H 2 О). Соединения железа (II), как правило, окисляются в воздухе до соединений железа (III).
Этимология
 «ирен», древнеанглийское слово, означающее «железо»
«ирен», древнеанглийское слово, означающее «железо»
Время железо использовалось так давно у него много имен. Источником его химического символа Fe является латинское слово ferrum, а его потомками являются названия элемента в романских языках (например, французский fer, испанский Иерро и итальянский и португальский ферро). Само слово ferrum, возможно, происходит от семитских языков через этрусский, от корня, который также начало дал древнеанглийскому bræs «латунь «. Английское слово железо происходит от протогерманского * isarnan, которое также является немецким именем Eisen. Скорее всего, оно было заимствовано из кельтского * isarnon, которое в конечном итоге происходит от протоиндоевропейского * is- (e) ro- «могущественный, святой» и, наконец, * eis « Клюге относит * isarnon к иллирийскому и латинскому ira, «гнев»). балто-славянские названия железа (например, русское железо [zhelezo], польское żelazo, литовское geležis) — единственные, которые происходят непосредственно от протоиндоевропейского * гельг — «железо». Китайский tiě (тип 鐵; упрощенный <658). также обозначения обозначения других предметов, сделанных из железа или стали, или, сделанных, из-за твердости и прочности металла.>铁) происходит от прото-сино-тибетского * hliek и был заимствован в Японский as 鉄 tetsu, в котором также есть местное чтение курогане «черный мета лл »(аналогично тому, как железо регистрируется в английском слове кузнец ).
История
Развитие металлургии железа
Железо — один из элементов, несомненно известному древнему миру. Его обрабатывали или ковали на протяжении тысячелетий. Однако железные предметы большого возраста встречаются гораздо реже, чем предметы из золота. или серебро из-за легкости, с которым железо разъедает. Технология развивалась медленно, и даже после открытия плавки потребовалось много веков, чтобы железо заменило бронзу в качестве металла, предпочитаемое для инструментов и оружия.
Метеоритное железо
Головка железного гарпуна из Гренландии. Железный край покрывает нарвал клык гарпуна, изготовленный из метеоритного железа из метеорита Кейп-Йорк, одного из ведущих иро Известно н метеоритов.
Бусы, сделанные из метеоритного железа в 3500 г. до н.э. или ранее были найдены в Герце, Египет, Г.А. Уэйнрайт. Бусины содержат 7,5% никеля, что является признаком метеорного происхождения, поскольку железо, обнаруженное происхождение в земной коре, обычно содержит незначительные примеси никеля.
Метеоритное железо высоко ценилось из-за своего происхождения небесного и часто использовалось для изготовления оружия и инструментов. Например, в гробнице Тутанхамона был найден кинжал, сделанный из метеоритного железа, созданный же пропорции железа, кобальта и никеля, что и обнаруженный в этом районе метеорит, отложенный древний метеоритный дождь. Данные, которые, вероятно, были сделаны египтянами из железа, датируются периодом от 3000 до 2500 до н.э.
Метеоритное железо сравнительно мягкое и пластичное, его легко ковать в холодном состоянии, но при нагревании оно может стать хрупким из-за содержанием никеля.
Кованое железо
 Символ Марса использовался с древних времен для обозначения железа. железный столб Дели примером методологий производительности и обработки железа в ранней Индии.
Символ Марса использовался с древних времен для обозначения железа. железный столб Дели примером методологий производительности и обработки железа в ранней Индии.
Первое производство железа началось в Среднем бронзовом веке, но несколько столетий, прежде чем железо вытеснило бронзу. Образцы выплавленного железа из Асмара, Месопотамии и Высокого Чагарского базара на севере Сирии были изготовлены где-то между 3000 и 2700 годами до нашей эры. Хетты основали империю в северо-центральной части Анатолии около 1600 г. до н.э. Похоже, они первыми осознали производство железа из руд и высоко оценили его в своем обществе. Хетты начали плавить железо между 1500 и 1200 годами до нашей эры, и эта практика распространилась на остальной Ближнем Востоке после падения их империи в 1180 году до нашей эры. Последующий период называется железным веком.
Артефакты плавленого железа обнаружены в Индии, датируемые 1800–1200 гг. До н.э., и в Леванте примерно с 1500 г. до н.э. (предполагаемая выплавку в Анатолии или Кавказе ). Предполагаемые ссылки (ср. история металлургии в Азии ) на железо в индийских Ведах использовались для очень раннем использовании железа в Индии, соответственно, чтобы датировать тексты как таковые.. ригведа термин аяс (металл), вероятно, относится к меди и бронзе, тогда как железо или śyāma ayas, впервые упоминается в постригведическом Атхарваведе.
Некоторые археологические свидетельства предполагает, что железо выплавляли в Зимбабве и в юго-восточной Африке еще в восьмом веке нашей эры. Обработка железа была завезена в Грецию в конце 11 века до н.э., откуда она быстро распространилась по Европе.
Железный серп из Древней Греции.
Распространение обработки железа в Центральной Европе связано с расширением кельтской. Согласно Плинию Старшему, железо было обычным делом в римскую эпоху. Годовое производство железа в Римской империи оценивается в 84750 т, в то время как в таком же густонаселенном и современном ханьском Китае производилось около 5000 тонн. В Китае железо появляется только около 700–500 лет до нашей эры. Выплавка могла быть завезена в Китай через Среднюю Азию. Самые ранние свидетельства использования доменной печи в Китае к I веку нашей эры, а вагранки использовались еще в период Вою царств (403–221 до н.э.). Использование дома и вагранок оставалось широко распространенных во время династий Сун и Тан.
. Во время промышленной революции в Великобритании Генри Корт начал рафинировать железо из свиней. от железа до кованого железа (или пруткового железа) с использованием инновационных производственных систем. В 1783 году он запатентовал процесс лужения для очистки железной руды. Позже он был улучшен другими, в том числе Джозеф Холл.
Чугун
Чугун был впервые произведен в Китае в 5 веке до нашей эры, но вряд ли был в Европе до нашей эры. средневековья. период. Самые ранние чугунные артефакты были обнаружены археологами на территории современного уезда Лухэ, Цзянсу в Китае. Чугун использовался в древнем Китае для ведения войны, сельского хозяйства и архитектуры. В период средневековья в Европе были найдены средства производства кованого железа из чугуна (в данном известном как чугун ) с использованием кузниц для украшений. Для всех этих процессов древесный уголь требовался в качестве топлива.
Coalbrookdale by Night, 1801. Доменные печи освещают металлургический город Coalbrookdale.
средневековые доменные печи. было около 10 футов (3,0 м) в высоту и выполнены из несгораемого кирпича; принудительный воздух обычно подавался сильфонами с ручным управлением. Современные доменные печи стали намного больше, с подами диаметром четырнадцать метров, что позволяет им выполнять тысячи тонн железа каждый день, по сути, они работают почти так же, как и в средневековые времена.
В 1709 году, Авраам Дарби I установил коксовую доменную печь для производства чугуна, заменив древесный уголь, но продолжая использовать доменные печи. Последовавшая доступность недорогого железа была одним из факторов, приведших к промышленной революции. К концу 18 века чугун начал заменять кованое железо для определенных целей, потому что он был дешевле. Содержание углерода в железе не считалось причиной различий в свойствах кованого железа, чугуна и стали до 18 века.
Оно становилось все более дешевым и доступным. Материал, из которого был построен инновационный первый железный мост в 1778 году. Этот мост до сих пор стоит в качестве памятника роли в промышленной революции. После этого железо использовалось в рельсах, лодках, кораблях, акведуках и зданиях, а также в железных цилиндрах в паровых машинах. Железные дороги сыграли центральную роль в формировании современности и идей прогресса, и на разных языках (например, французском, испанском, итальянском и немецком) железные дороги называются железной дорогой.
Сталь
Сталь (с меньшим содержанием углерода, чем чугун, но большим, чем у кованого железа) впервые была произведена в древности с использованием блюмера. Кузнецы Луристана в западной Персии делали хорошую сталь к 1000 году до нашей эры. Затем около 300 г. до н.э. и 500 г. н.э., были разработаны улучшенные версии, сталь Wootz в Индии и дамасская сталь. Эти методы были специализированными, поэтому сталь не стала основным товаром до 1850-х годов.
Новые методы ее производства науглероживания железных стержней в процессе цементирования были изобретены в 17 веке. Во время промышленной революции были изобретены новые методы производства пруткового железа без древесного угля, которые позже были применены для производства стали. В конце 1850-х годов Генри Бессемер изобрел новый процесс стали, предусматривающий продувку воздуха стали через расплавленный чугун для производства мягкой стали. Это сделало сталь намного более экономичной, что привело к тому, что кованое железо больше не производилось в больших количествах.
Основы современной химии
В 1774 году Антуан Лавуазье реакция водяного пара с металлическим железом внутри раскаленной железной трубки для производства водорода в его экспериментах, чтобы привести к демонстрации чтобы сыграть роль в превращении химии из качественной науки в количественный.
Символическая роль
«Gold gab ich für Eisen» — «Я отдал золото за железо». Немецко-американская брошь времен Первой мировой войны.
Железо играет определенную роль в мифологии и нашем различном использовании как метафора и в фольклоре. Греческий поэт Гесиод Труды и дни (строки 109–201) перечисляет разные возрасты человека, названные в честь металлов, таких как золото, серебро, бронза и железо, чтобы преследовать последовательные эпохи человечества. Железный век был связан с Римом, и в «Метаморфозах» Овидия
Добродетели в отчаянии покинули землю; и порочность человека становится всеобщей и полной. Тогда на смену пришла твердая сталь.
— Овидий, Метаморфозы, Книга I, Железный век, строка 160 и далее
Пример важности символической роли железа можно найти в Немецкой кампании 1813. Фридрих Вильгельм III заказал тогда первый Железный крест в качестве военной награды. Берлинские железные украшения достигли пика производства между 1813 и 1815 годами, когда прусская королевская семья призвала граждан жертвовать золотые и серебряные украшения на военные нужды. Надпись Gold gab ich für Eisen (Я дал золото за железо) также использовалась в более поздних военных усилиях.
Производство металлического железа
Железный порошок
Лабораторные маршруты
Для нескольких ограниченных Когда это необходимо, чистое железо производится в лаборатории в небольших количествах, восстанавливая чистый оксид или гидроксид водородом, или образуя пентакарбонил железа и нагревая его до 250 ° C, чтобы оно разлагалось с образованием чистого порошка железа. Другой метод — электролиз хлористого железа на железном катоде.
Основной производственный маршрут
| Страна | Железная руда | Чугун | Прямое железо | Сталь |
|---|---|---|---|---|
| Китай | 1,114,9 | 549,4 | 573,6 | |
| Австралия | 393,9 | 4,4 | 5,2 | |
| Бразилия | 305.0 | 25.1 | 0.011 | 26,5 |
| Япония | 66,9 | 87,5 | ||
| Индия | 257,4 | 38,2 | 23,4 | 63,5 |
| Россия | 92,1 | 43,9 | 4,7 | 60,0 |
| Украина | 65,8 | 25,7 | 29,9 | |
| Южная Корея | 0,1 | 27,3 | 48,6 | |
| Германия | 0,4 | 20,1 | 0,38 | 32,7 |
| Мир | 1,594,9 | 914,0 | 64,5 | 1,232,4 |
В настоящее время промышленное производство чугуна или стали состоит из двух основных этапов. На первом этапе железная руда восстанавливается с коксом в доменной печи, а расплавленный металл отделяют от грубых примесей, таких как силикатные мин ералы. На этой стадии получают сплав чугун, который содержит относительно большие количества углерода. На втором этапе количество углерода в передельном чугуне снижается за счет окисления с получением кованого железа, стали или чугуна. На этом этапе могут быть добавлены другие металлы для образования легированных сталей.
Китайское изображение рабочих доменной печи, 17 век, изготовление кованого железа из чугуна Как добывали железо в XIX веке
Взрыв обработка печи
Доменная печь загружается железной рудой, обычно гематитом Fe. 2O. 3 или магнетитом Fe. 3O. 4, вместе с коксом (уголь, который был отдельно запекается для удаления летучих компонентов). Воздух, предварительно нагретый до 900 ° C, пропускается через смесь в количестве, достаточном для превращения углерода в монооксид углерода :
- 2 C + O 2 → 2 CO
В этой реакции возникает температура около 2000 ° C. Монооксид углерода восстанавливает железную руду до металлического железа
- Fe2O3+ 3 CO → 2 Fe + 3 CO 2
Некоторое количество железа в высокотемпературной нижней области печи вступает в реакцию непосредственно с коксом:
- 2 Fe 2O3+ 3 C → 4 Fe + 3 CO 2
A флюс, например, известняк (карбонат кальция ) или доломит (карбонат кальция-магния) также добавляется к загрузке печи. Его цель — удалить из руды кремнеземистые минералы, которые в противном случае засорили бы печь. Тепло печи разлагает карбонаты до оксида кальция, который реагирует с любым избытком кремнезема с образованием шлака, состоящего из силиката кальция. CaSiO. 3 или другие продукты. При температуре печи и металл, и шлак расплавляются. Они собираются внизу в виде двух несмешивающихся жидких слоев (со шлаком наверху), которые затем легко разделяются. Шлак можно использовать в качестве материала в строительстве дорог или для улучшения бедных минералами почв для сельского хозяйства.
Эта куча железорудных окатышей будет использоваться в производстве стали.
Производство стали
Котел с расплавленным чугуном, который используется для производства стали
Как правило, чугун, полученный в доменной печи, содержит до 4–5% углерода с небольшими количествами других примесей, таких как сера, магний, фосфор и марганец. Высокое содержание углерода делает его относительно непрочным и хрупким. Уменьшение количества углерода до 0,002–2,1% по массе дает сталь , которая может быть в 1000 раз тверже чистого железа. Затем можно изготавливать большое количество различных стальных изделий с помощью холодной обработки, горячей прокатки, ковки, механической обработки и т. Д. Удаление других примеси, напротив, приводят к получению чугуна, который используется для литья изделий в литейных цехах ; например, печи, трубы, радиаторы, фонарные столбы и рельсы.
Стальные изделия часто подвергаются различной термообработке после того, как они кованы для придания формы. Отжиг заключается в нагреве до 700–800 ° C в течение нескольких часов с последующим постепенным охлаждением. Это делает сталь более мягкой и податливой.
Прямое восстановление железа
Из-за экологических проблем были разработаны альтернативные методы обработки железа. «Прямое восстановление железа » восстанавливает железную руду до куска железа, называемого «губчатое» железо или «прямое» железо, которое подходит для выплавки стали. Процесс прямого восстановления включает две основные реакции:
Природный газ частично окисляется (с теплом и катализатором):
- 2 CH 4 + O 2 → 2 CO + 4 H 2
Затем этими газами в печи обрабатывают железную руду, получая твердое губчатое железо:
- Fe2O3+ CO + 2 H 2 → 2 Fe + CO 2 + 2 H 2O
Кремнезем удаляется добавлением флюса известняка, как описано выше.
Thermite process
Воспламенение смеси алюминиевого порошка и оксид железа дает металлическое железо посредством термитной реакции :
- Fe2O3+ 2 Al → 2 Fe + Al 2O3
В качестве альтернативы передельный чугун может быть превращен в сталь (примерно с 2% углерода) или кованое железо (технически чистое железо). Для этого использовались различные процессы, в том числе кузницы для отделки, пудлинговые печи, конвертеры Бессемера, мартеновские печи, основные кислородные печи и дуговые электропечи. Во всех случаях цель состоит в том, чтобы окислить часть или весь углерод вместе с другими примесями. С другой стороны, другие металлы могут быть добавлены для производства легированных сталей.
Области применения
В качестве конструкционного материала
Железо является наиболее широко используемым из всех металлов, что составляет более 90% мирового производства металла. Его низкая стоимость и высокая прочность часто делают его предпочтительным материалом для выдерживания напряжений или передачи сил, таких как конструкция машин и станков, рельсов, автомобилей, корпуса судов, железобетонная арматура и несущий каркас зданий. Поскольку чистое железо довольно мягкое, его чаще всего сочетают с легирующими элементами для получения стали.
Механические свойства
| Материал | TS. (МПа) | BH. (Brinell ) |
|---|---|---|
| усы железа | 11000 | |
| Аусформированная (закаленная). сталь | 2930 | 850–1200 |
| Мартенситная сталь | 2070 | 600 |
| бейнитная сталь | 1380 | 400 |
| Перлитная сталь | 1200 | 350 |
| Холоднодеформированное железо | 690 | 200 |
| Мелкозернистое железо | 340 | 100 |
| Углеродосодержащее железо | 140 | 40 |
| Чистое, простое -кристаллическое железо | 10 | 3 |
Механические свойства железа и его сплавов чрезвычайно важны для их структурного применения. Эти свойства можно оценивать различными способами, включая тест Бринелля, тест Роквелла и й e Испытание на твердость по Виккерсу.
Свойства чистого железа часто используются для калибровки измерений или сравнения результатов испытаний. Однако на механические свойства железа в значительной степени влияет чистота образца: чистые монокристаллы железа на самом деле мягче алюминия, а самое чистое промышленно производимое железо (99,99%) имеет твердость 20–30 по Бринеллю.
Увеличение содержания углерода приведет к значительному увеличению твердости и прочности чугуна. Максимальная твердость 65 R c достигается при содержании углерода 0,6%, хотя сплав имеет низкую прочность на разрыв. Из-за мягкости железа работать с ним намного легче, чем с его более тяжелыми конгенераторами рутением и осмием.
Диаграмма состояния железо-углерод
Типы сталей и сплавы
α-Железо — довольно мягкий металл, способный растворять лишь небольшую концентрацию углерода (не более 0,021% по массе при 910 ° C). Аустенит (γ-железо) такой же мягкий и металлический, но может растворять значительно больше углерода (до 2,04% по массе при 1146 ° C). Эта форма железа используется в типе нержавеющей стали, используемой для изготовления столовых приборов, а также оборудования для больниц и предприятий общественного питания.
Коммерчески доступное железо классифицируется по чистоте и количеству добавок.. Чугун содержит 3,5–4,5% углерода и содержит различные количества загрязняющих веществ, таких как сера, кремний и фосфор. Чугун — это не товарный продукт, а скорее промежуточный этап в производстве чугуна и стали. Уменьшение содержания загрязняющих веществ в передельном чугуне, которые негативно влияют на свойства материала, таких как сера и фосфор, дает чугун, содержащий 2–4% углерода, 1–6% кремния и небольшое количество марганца. Чугун имеет температуру плавления в диапазоне 1420–1470 К, что ниже, чем у любого из двух его основных компонентов, и делает его первым продуктом, плавящимся при совместном нагревании углерода и железа. Его механические свойства сильно различаются и зависят от формы, которую углерод принимает в сплаве.
«Белые» чугуны содержат углерод в форме цементита или карбида железа (Fe 3 С). Этот твердый, хрупкий состав доминирует в механических свойствах белого чугуна, делая его твердым, но неустойчивым к ударам. Изломанная поверхность белого чугуна полна мелких граней битого карбида железа, очень бледного, серебристого, блестящего материала, отсюда и название. При медленном охлаждении смеси железа с 0,8% углерода ниже 723 ° C до комнатной температуры образуются отдельные чередующиеся слои цементита и α-железа, которое является мягким и податливым и называется перлит из-за его внешнего вида. С другой стороны, быстрое охлаждение не оставляет времени для этого разделения и создает твердый и хрупкий мартенсит. Затем сталь может быть подвергнута отпуску путем повторного нагрева до промежуточной температуры, изменяя пропорции перлита и мартенсита. Конечный продукт с содержанием углерода ниже 0,8% представляет собой смесь перлит-αFe, а содержание углерода выше 0,8% представляет собой смесь перлит-цементит.
В сером чугуне углерод существует отдельно, мелкие хлопья графита, а также делают материал хрупким из-за чешуек графита с острыми краями, которые создают участки концентрации напряжений внутри материала. Более новый вариант серого чугуна, именуемый высокопрочный чугун, специально обрабатывают следовыми количествами магния, чтобы изменить форму графита на сфероиды или конкреции, снижая концентрации напряжений и значительно увеличивает ударную вязкость и прочность материала.
Кованое железо содержит менее 0,25% углерода, но большое количество шлака придает ему волокнистость. Это прочный, податливый продукт, но не такой плавкий, как чугун. Если заточить до края, он быстро его теряет. Кованое железо характеризуется наличием тонких волокон шлака, заключенных в металле. Кованое железо более устойчиво к коррозии, чем сталь. Ее почти полностью заменили на низкоуглеродистую сталь для традиционных изделий из «кованого железа» и кузнечное дело.
Мягкая сталь подвержена коррозии быстрее, чем кованое железо, но она дешевле и более доступна. Углеродистая сталь содержит 2,0% углерода или менее, с небольшими количествами марганца, серы, фосфора и кремния. Легированные стали содержат различные количества углерода, а также других металлов, таких как хром, ванадий, молибден, никель, вольфрам и т. д. Содержание в них сплава увеличивает их стоимость, поэтому они обычно используются только для специальных целей. Однако наиболее распространенной легированной сталью является нержавеющая сталь. Последние разработки в черной металлургии привели к производству все большего количества микролегированных сталей, также называемых «HSLA », или высокопрочных, низколегированных сталей, содержащих крошечные добавки, обеспечивающие высокую прочность и часто впечатляющую ударную вязкость при минимальных затратах.
Фотон массовый коэффициент ослабления для железа.
Помимо традиционных применений, железо также используется для защиты от ионизирующего излучения. Хотя он легче другого традиционного защитного материала, свинца, он намного прочнее механически. На графике показано ослабление излучения в зависимости от энергии.
Главный недостаток чугуна и стали заключается в том, что чистое железо и большинство его сплавов сильно страдают от ржавчины, если не защищены каким-либо образом, затраты составляют более 1% мировой экономики. Покраска, гальванизация, пассивация, пластиковое покрытие и воронение используются для защиты железа от ржавчины за счет исключения воды и кислорода или с помощью катодной защиты. Механизм ржавчины железа следующий:
- Катод: 3 O 2 + 6 H 2 O + 12 e → 12 OH
- Анод: 4 Fe → 4 Fe + 8 e; 4 Fe → 4 Fe + 4 e
- Всего: 4 Fe + 3 O 2 + 6 H 2 O → 4 Fe + 12 OH → 4 Fe (OH) 3 или 4 FeO (OH) + 4 H 2O
Электролит обычно сульфат железа (II) в городских районах (образуется при атмосферном диоксиде серы атакует железо) и частицы соли в атмосфере в приморских районах.
Соединения железа
Хотя в основном железо используется в металлургии, соединения железа также широко распространены в промышленности. Железные катализаторы традиционно используются в процессе Хабера-Боша для производства аммиака и процессе Фишера-Тропша для превращения монооксида углерода в углеводороды для топлива и смазочные материалы. Порошковое железо в кислотном растворителе использовалось в восстановлении Бешампа восстановлении нитробензола до анилина.
оксида железа (III), смешанного с алюминием. Порошок может быть воспламенен для создания реакции термитов, используемой при сварке больших железных деталей (например, рельсов ) и очистке руды. Оксид железа (III) и используется как красноватый и охристый пигменты.
Хлорид железа (III) находит применение при очистке воды и очистке сточных вод, при крашении ткани, в качестве краситель в красках, в качестве добавки к корму для животных и в качестве травителя для меди при производстве печатных плат. Его также можно растворить в спирте с образованием настойки железа, которая используется в качестве лекарства для остановки кровотечения у канареек.
Сульфат железа (II) используется в качестве предшественника других соединений железа. Он также используется для восстановления хромата в цементе. Он используется для обогащения пищевых продуктов и лечения железодефицитной анемии. Сульфат железа (III) используется для осаждения мельчайших частиц сточных вод в баковой воде. Хлорид железа (II) используется в качестве восстанавливающего флокулянта, при образовании комплексов железа и магнитных оксидов железа, а также в качестве восстановителя в органическом синтезе.
Биологическая и патологическая роль
Железо необходимо для жизни. Кластеры железо-сера распространены и включают нитрогеназу, ферменты, ответственные за биологическую фиксацию азота. Железосодержащие белки участвуют в транспортировке, хранении и использовании кислорода. Белки железа участвуют в переносе электрона.
Структура гема b ; в белке дополнительный лиганд (ы) может быть присоединен к Fe.
Примеры железосодержащих белков у высших организмов включают гемоглобин, цитохром (см. высокомалентный железо ) и каталаза. Средний взрослый человек содержит около 0,005% веса тела железа, или около четырех граммов, из которых три четверти приходится на гемоглобин — уровень, который остается постоянным, несмотря на то, что каждый день усваивается только около одного миллиграмма железа, поскольку человеческое тело перерабатывает свой гемоглобин для содержания железа.
Биохимия
Получение железа представляет проблему для аэробных организмов, поскольку трехвалентное железо плохо растворяется вблизи нейтрального pH. Таким образом, эти организмы разработали средства для поглощения железа в виде комплексов, иногда поглощая двухвалентное железо, прежде чем окислять его обратно до трехвалентного железа. В частности, бактерии развили очень высокоаффинные секвестрирующие агенты, называемые сидерофорами.
. После поглощения человеческими клетками хранение железа точно регулируется. Основным компонентом этой регуляции является белок трансферрин, который связывает ионы железа, абсорбированные из двенадцатиперстной кишки, и переносит его с кровью в клетки. Трансферрин содержит Fe в центре искаженного октаэдра, связанный с одним азотом, тремя атомами кислорода и хелатирующим карбонатным анионом, который захватывает ион Fe: у него такая высокая константа стабильности , что он очень эффективно поглощает ионы Fe даже из самых стабильных комплексов. В костном мозге трансферрин восстанавливается из Fe и Fe и сохраняется в виде ферритина для включения в гемоглобин.
Наиболее известные и изученные биоинорганические соединения железа ( биологические молекулы железа) представляют собой гемовые белки : примерами являются гемоглобин, миоглобин и цитохром P450. Эти соединения участвуют в транспортировке газов, создании ферментов и переносе электронов. Металлопротеины представляют собой группу белков с кофакторами ионов металлов. Некоторыми примерами металлопротеинов железа являются ферритин и рубредоксин. Многие ферменты, жизненно важные для жизни, содержат железо, такие как каталаза, липоксигеназы и IRE-BP.
Гемоглобин — переносчик кислорода, который содержится в красных кровяных тельцах. и способствует их окраске, транспортируя кислород по артериям от легких к мышцам, где он передается в миоглобин, который сохраняет его до тех пор, пока он не понадобится для метаболического окисления глюкозы, генерируя энергию. Здесь гемоглобин связывается с углекислым газом, образующимся при окислении глюкозы, который транспортируется по венам гемоглобином (преимущественно в виде анионов бикарбоната ) обратно в легкие, где он выдыхается. В гемоглобине железо находится в одной из четырех групп гема и имеет шесть возможных координационных центров; четыре заняты атомами азота в кольце порфирина, пятое — атомом азота имидазола в остатке гистидина одной из белковых цепей, присоединенной к гемовой группе, а шестой зарезервирован для молекулы кислорода, с которой он может обратимо связываться. Когда гемоглобин не присоединен к кислороду (и тогда его называют дезоксигемоглобином), ион Fe в центре группы гема (внутри гидрофобного белка) находится в высокоспиновой конфигурации. Таким образом, оно слишком велико, чтобы поместиться внутри порфиринового кольца, которое вместо этого изгибается в купол с ионом Fe примерно на 55 пикометров над ним. В этой конфигурации шестой координационный центр, зарезервированный для кислорода, заблокирован другим остатком гистидина.
Когда дезоксигемоглобин захватывает молекулу кислорода, этот остаток гистидина удаляется и возвращается, как только кислород надежно присоединяется, образуя водородная связь с ним. Это приводит к переключению иона Fe на низкоспиновую конфигурацию, что приводит к уменьшению ионного радиуса на 20%, так что теперь он может помещаться в порфириновое кольцо, которое становится плоским. (Кроме того, эта водородная связь приводит к наклону молекулы кислорода, в результате чего валентный угол Fe — O — O составляет около 120 °, что позволяет избежать образования мостиков Fe — O — Fe или Fe — O 2 –Fe Это приводит к перемещению всех белковых цепей, что приводит к изменению других форм субъединиц гемоглобина на форму с большим сродством к кислороду. Таким образом, когда дезоксигемоглобин поглощает кислород, его слияние к большему количеству кислорода увеличивается, и наоборот. Миоглобин, с другой стороны, содержит только одну гемовую группу, и, следовательно, этот кооперативный эффект не может возникнуть. Таким образом, хотя гемоглобин почти насыщен кислородом при высоких парциальных давлениях кислорода в легких, его сродство к кислороду намного, чем у миоглобина, который насыщает кислородом даже при низких парциальных давлениях кислорода в мышечной ткани. Как описано с помощью эффект Бора (названного в честь Кристиана Бора, отца Нильса Бора ), сродство гемоглобина к кислороду уменьшено в приведенном диоксида углерода.
Гемовая единица человеческого карбоксигемоглобина, показывающая карбонильный лиганд в апикальном положении, транс к остатку гистидина.
Окись углерода и фосфор Трифторид ядовит для человека, потому что он связан с гемоглобином так же, как с кислородом, но с гораздо большей силой, так что кислород больше не может переноситься по всему телу. Гемоглобин, связанный с оксидом углерода, известен как карбоксигемоглобин. Этот эффект также играет второстепенную роль в токсичности цианида, но главный эффектом является его вмешательство в правильное функционирование белка-транспортера электронов цитохрома А. Белки цитохрома также включают гемовые группы и участвуют в метаболическом окислении глюкозы кислородом. Затем этот шестой координационный сайт занимает либо другой имидазольный азот, либо метионин сера, так что эти белки в степени инертны по отношению к кислороду — за исключением цитохрома a, который напрямую связывается с кислородом и, таким образом, очень легко отравлен цианидом. Здесь перенос электрона происходит, когда железо остается в низком спине, но изменяется между состояниями окисления +2 и +3. Возможности восстановления на каждом этапе немного больше, чем на предыдущем, энергия постепенно увеличивается и таким образом, может храниться в аденозинтрифосфате. Цитохром немного отличается, как он находится на митохондриальной мембране, связывается с кислородом и переносит протоны, а также электроны следующим образом:
- 4 Cytc + O 2 + 8H. внутри → 4 Cytc + 2 H 2 O + 4H. снаружи
Хотя гемовые белки являются наиболее важными классом железосодержащих белков, белки железо-сера также очень важны, поскольку они участвуют в переносе электронов, что возможно, поскольку железо может стабильно существовать в состоянии окисления +2 или +3. Они имеют, два, четыре или четыре атома железа, каждый из которых представляет собой тетраэдрически координирован с четырьмя атомами серы; из-за этой тетраэдрической системы в них всегда есть высокоспиновое железо. Самым только из таких соединений является рубредоксин, который имеет один атом железа, координированный с четырьмя атомами серы от остатков цистеина в окружающих пептидных цепях. Другой важный класс железо-серных белков — это ферредоксины, которые имеют несколько атомов железа. Трансферрин не принадлежит ни к одному из этих классов.
Способность морских мидий удерживать свои позиции на скалах в океане обеспечивается за счет использования металлоорганического железа -основные связи в их богатой белком кутикуле. Основываясь на синтетических аналогах, обнаружение железа в этой структурех увеличивало модуль упругости в 770 раз, предел прочности на разрыв в 58 раз и ударную вязкость в 92 раза. Уровень стресса, необходимый для их необратимого повреждения, увеличился в 76 раз.
Питание
Диета
Железо широко распространено, но особенно богатые источники пищевого железа, включая красное мясо, устрицы, чечевица, фасоль, птица, рыба, листовые овощи, кресс-салат, тофу, нут, черноглазый горох и меласса. Хлеб и хлопья для завтрака иногда специально обогащены железом.
Железо, содержащееся в пищевых добавках, часто встречается в виде фумарата железа (II), хотя сульфат железа (II) дешевле и одинаково хорошо абсорбируется. Элементарное железо или восстановленное железо, несмотря на то, что абсорбируется на уровне одной трети до двух третей эффективности (по сравнению с сульфатом железа), часто в такие продукты, как сухие завтраки или обогащенная пшеничная мука. Железо наиболее доступно для организма, когда хелатировано с аминокислотами, а также доступно для использования в качестве обычной добавки железа. Глицин, наименее дорогая аминокислота, чаще всего используется для производства добавок глицината железа.
по Рекомендации питанию
Институт медицины США (IOM) обновил средние расчетные потребности (EAR) и Рекомендуемая диетическая норма (RDA) для железа в 2001 году. Текущий EAR для железа для женщин в возрасте 14–18 лет составляет 7,9 мг / день, 8,1 мг / день для возраста 19–50 лет и 5,0 мг в последующий период (после менопаузы). Для мужчин EAR составляет 6,0 мг / день в возрасте от 19 лет и старше. Рекомендуемая суточная норма составляет 15,0 мг / день для женщин в возрасте 15–18 лет, 18,0 мг для женщин 19–50 лет и 8,0 в дальнейшем. Для мужчин: 8,0 мг / день в возрасте от 19 лет и старше. RDA выше, чем EAR, чтобы определить сумму, которая покроют людей с потребностями выше среднего. Рекомендуемая суточная доза для беременных составляет 27 мг / день, а для кормления грудью — 9 мг / день. Для детей в возрасте 1-3 лет — 7 мг / день, 10 — в возрасте 4-8 лет и 8 — в возрасте 9-13 лет. Что касается безопасности, IOM также устанавливает допустимые верхние уровни потребления (UL) для витаминов и минералов, если доказательств достаточно. В случае железа верхний предел установлен на уровне 45 мг / день. В совокупности EAR, RDA и UL обозначаются как Референтное потребление пищи.
Европейское управление по безопасности пищевых продуктов (EFSA) называемыми совокупным набором информации контрольными значениями питания с эталонным потреблением населения. (PRI) вместо RDA и Среднее требование вместо EAR. AI и UL: экологически чистая продукция так же, как в США. Для женщин PRI составляет 13 мг / день в возрасте 15–17 лет, 16 мг / день для женщин в возрасте 18 лет и старше в пременопаузе и 11 мг / день в постменопаузе. При беременности и кормлении грудью — 16 мг / сут. Для мужчин PRI составляет 11 мг / день в возрасте 15 лет и старше. Для детей в возрасте от 1 до 14 лет PRI увеличивается с 7 до 11 мг / день. PRI выше, чем RDA в США, за исключением беременности. EFSA рассмотрело тот же вопрос безопасности и не установило UL.
Младенцам могут потребляться добавки железа, если они кормятся из коровьего молока из бутылочки. Часто доноры крови подвержены риску низкого уровня железа, и им рекомендуют потребление железа.
Для целей маркировки пищевых продуктов и пищевых добавок в США количество в порции выражается в процентах дневной стоимости (% DV). Для целей маркировки железа 100% дневной нормы составляющего 18 мг, а по состоянию на 27 мая 2016 г. оставалось оставшимся на уровне 18 мг. Соблюдение обновленных правил маркировки требовалось к 1 января 2020 года для производителей с годовым объемом продаж продуктов питания 10 миллионов долларов и более и к 1 января 2021 года для производителей с годовым объемом продаж продуктов питания менее миллионов долларов. В течение первых шести месяцев после даты соответствия 1 января 2020 года FDA сотрудничает с новыми требованиями к этикеткам Nutrition Facts. Таблица старых и новых суточных значений для взрослых представлена в разделе Референсная суточная доза.
Дефицит
Дефицит железа является наиболее распространенным дефицитом питания в мире. Когда железа не компенсируется адекватным потреблением железа с пищей, возникает состояние латентного дефицита железа, которое со временем приводит к железодефицитной анемии, если ее не лечить, что проявляется недостаточным эритроцитов и недостаточным количеством гемоглобина. Наиболее подвержены заболеванию дети, пременопаузальные женщины (женщины детородного возраста) и люди с плохим питанием. В большинстве случаев железодефицитная анемия протекает в легкой форме.
Избыток
Поглощение железа строго регулируется человеческим телом, у которого нет регулируемых физиологических средств выделения железа. Ежедневно теряется лишь небольшое количество железа из-за слущивания эпителиальных клеток слизистой оболочки, оболочек и кожи, контроль уровня железа в основном достигается за счет регулирования поглощения. Регуляция поглощения железа у некоторых людей нарушена в результате генетического дефекта, который отображается в области гена HLA-H на хромосоме 6 и приводит к аномально низким уровням гепсидин, ключевой регулятор поступления железа в систему кровообращения у млекопитающих. У этих людей чрезмерное потребление железа может привести к расстройствам, способным вызвать перегрузку железом, которые в медицине известны как гемохроматоз. Многие люди имеют не диагностированную генетическую предрасположенность к перегрузке железом и не знают семейного анамнеза этой проблемы. По этой причине людям не следует принимать добавки железа, если они не страдают дефицитом железа и не проконсультировались с врачом. Гемохроматоз считается от 0,3 до 0,8% всех метаболических заболеваний у европеоидов.
Передозировка проглоченного железа может вызвать чрезмерный уровень свободного железа в крови. Высокое содержание свободного двухвалентного железа в крови реагирует с пероксидами с образованием высокореактивных свободных радикалов, которые могут повредить ДНК, белки, липиды и другие клеточные компоненты. Проблемы возникают, когда возникают проблемы, когда возникают проблемы, когда возникают уровни железа, превышающие доступность трансферрина для связывания железа. Повреждение клеток желудочно-кишечного тракта также может помешать им регулировать абсорбцию железа, что приведет к дальнейшему повышению уровня в крови. Железо обычно повреждает клетки в сердце, печени и других местах, вызывая побочные эффекты, включая кому, метаболический ацидоз, шок., печеночная недостаточность, коагулопатия, респираторный дистресс-синдром у взрослых, длительное повреждение органов и даже смерть. Люди испытывают отравление железом, когда содержание железа превышает 20 миллиграммов на каждый килограмм массы тела; 60 миллиграммов на килограмм считаются смертельной дозой. Чрезмерное потребление железа, часто используемое в результате употребления большого количества таблеток сульфата железа, предназначенное для употребления взрослыми, одним из наиболее частых токсикологических причин смерти в возрасте до шести лет. Референсное потребление с пищей (DRI) устанавливает максимально допустимый уровень потребления (UL) для взрослых на уровне 45 мг / день. Для детей младше четырнадцати лет UL составляет 40 мг / день.
Медицинское лечение отравления железом является сложным и дополнительным использованием специального хелатирующего агента, называемого дефероксамином связывать и выводить излишки железа из организма.
Рак
Роль железа в защите от рака может быть описана как «палка о двух концах» из-за его повсеместного присутствия в не -патологических процессах. У людей, проходящих химиотерапия, может развиться дефицит железа и анемия, при которых внутривенная терапия железом используется для восстановления уровня железа. Перегрузка железом, которая может вызвать рост опухоли и повысить предрасположенность к развитию рака, особенно колоректального рака.
См. Также
 Химический портал
Химический портал
- Эль-Мутун в Боливии, где находится 10% доступной в мире железной руды
- Наночастица железа
- Наночастица железо-платина
- Удобрение железом — удобено удобрение океанов для стимулирования фитопланктона рост
- железоокисляющие бактерии
- Список стран по производству железа
- окомкование — создание железорудных окатышей
- нержавеющее железо
- Сталь
- Железный цикл
Список литературы
Библиография
- Гринвуд, Норман Н. ; Эрншоу, Алан (1997). Химия элементов (2-е изд.). Баттерворт-Хайнеманн. ISBN 978-0-08-037941-8.
- Недели, Мэри Эльвира ; Лейчестер, Генри М. (1968). «Элементы, известные древним». Открытие элементов. Истон, Пенсильвания: Журнал химического образования. Стр. 29 –40. ISBN 0-7661-3872-0. LCCN 68-15217.
Дополнительная литература
- H.R. Шуберт, История британской черной металлургии… до 1775 года нашей эры (Рутледж, Лондон, 1957)
- Р.Ф. Тайлекот, История металлургии (Институт материалов, Лондон, 1992 г.).
- Р.Ф. Тайлекот, «Железо в промышленной революции» в J. Day и R.F. Тайлекот, Промышленная революция в металлах (Институт материалов, 1991), 200–60.
Внешние ссылки
| Найдите iron в Wiktionary, бесплатном формате. |
| Викискладе есть материалы, связанные с железом. |
- It’s Elemental — Железо
- Железо в Периодическая таблица видео (Ноттингемский университет)
- Металлургия для неметаллургов
- Железо Дж.Б. Калверт
| Железо | |
|---|---|
| Атомный номер | 26 |
| Внешний вид простого вещества |  ковкий, вязкий металл серебристо-белого цвета ковкий, вязкий металл серебристо-белого цвета |
| Свойства атома | |
| Атомная масса (молярная масса) | 55,847 а. е. м. (г/моль) |
| Радиус атома | 126 пм |
| Энергия ионизации (первый электрон) | 759,1 (7,87) кДж/моль (эВ) |
| Электронная конфигурация | [Ar] 3d6 4s2 |
| Химические свойства | |
| Ковалентный радиус | 117 пм |
| Радиус иона | (+3e) 64 (+2e) 74 пм |
| Электроотрицательность (по Полингу) | 1,83 |
| Электродный потенциал | Fe←Fe3+ −0,04 В Fe←Fe2+ −0,44 В |
| Степени окисления | 6, 3, 2, 0, −2 |
| Термодинамические свойства простого вещества | |
| Плотность | 7,874 г/см³ |
| Молярная теплоёмкость | 25,14[1] Дж/(K·моль) |
| Теплопроводность | 80,4 Вт/(м·K) |
| Температура плавления | 1812 K |
| Теплота плавления | 247,1 кДж/кг 13,8 кДж/моль |
| Температура кипения | 3134 K |
| Теплота испарения | ~6088 кДж/кг ~340 кДж/моль |
| Молярный объём | 7,1 см³/моль |
| Кристаллическая решётка простого вещества | |
| Структура решётки | кубическая объёмноцентрированная |
| Параметры решётки | 2,866 Å |
| Отношение c/a | — |
| Температура Дебая | 460 K |
| Fe | 26 |
| 55,847 | |
| [Ar]3d64s2 | |
| Железо |
Железо — элемент побочной подгруппы восьмой группы четвёртого периода периодической системы химических элементов Д. И. Менделеева, атомный номер 26. Обозначается символом Fe (Ferrum). Один из самых распространённых в земной коре металлов (второе место после алюминия).
Простое вещество железо (CAS-номер: 7439-89-6) — ковкий металл серебристо-белого цвета с высокой химической реакционной способностью: железо быстро корродирует при высоких температурах или при высокой влажности на воздухе. В чистом кислороде железо горит, а в мелкодисперсном состоянии самовозгорается и на воздухе.
На самом деле железом обычно называют его сплавы с малым содержанием примесей (до 0,8 %), которые сохраняют мягкость и пластичность чистого металла. Но на практике чаще применяются сплавы железа с углеродом: сталь (до 2 % углерода) и чугун (более 2 % углерода), а также нержавеющая (легированная) сталь с добавками легирующих металлов (хром, марганец, никель и др.). Совокупность специфических свойств железа и его сплавов делают его «металлом № 1» по важности для человека.
В природе железо редко встречается в чистом виде, чаще всего оно встречается в составе железо-никелевых метеоритов. Распространённость железа в земной коре — 4,65 % (4-е место после O, Si, Al). Считается также, что железо составляет бо́льшую часть земного ядра.
История
Железо как инструментальный материал известно с древнейших времён, самые древние изделия из железа, найденные при археологических раскопках, датируются 4-м тысячелетием до н. э. и относятся к древнешумерской и древнеегипетской цивилизациям. Это наконечники для стрел и украшения из метеоритного железа, то есть, сплава железа и никеля (содержание последнего колеблется от 5 до 30 %), из которого состоят метеориты. От их небесного происхождения идёт, видимо, одно из наименований железа в греческом языке: «сидер» (а на латыни это слово значит «звёздный»).
Изделия из железа, полученного искусственно, известны со времени расселения арийских племён из Европы в Азию и острова Средиземного моря (4—3-е тысячелетие до н. э.). Самый древний железный инструмент из известных — стальное долото, найденное в каменной кладке пирамиды Хеопса в Египте (построена около 2550 года до н. э.). Железо часто упоминается в древнейших (3-е тысячелетие до н. э.) текстах хеттов, основавших свою империю на территории современной Анатолии в Турции. Например, в тексте хеттского царя Анитты (около 1800 года до н. э.) говорится:
Когда на город Пурусханду в поход я пошел, человек из города Пурусханды ко мне поклониться пришел (…?) и он мне 1 железный трон и 1 железный скипетр (?) в знак покорности (?) преподнес.
В этом тексте железо обозначается словом «par-zi-lum» (сравните латинское «ferrum» и русское «железо»), что, скорее всего, значит «олово всадников» — от древнеарийских слов «PARSA» или «FERSY» (всадник — сравните этноним «персы», отсюда же шахматная фигура «ферзь», и латинские слова «persona» и «partia»), и корня «ZIL» (олово, и вообще белый металл).
В древности мастерами железных изделий слыли халибы, которых Геродот перечисляет в числе эллинских племён Малой Азии, подвластных Крезу. Халибы жили на севере державы Хеттов, у побережья Чёрного моря возле устья реки Галис (современный г. Самсун в Турции), и от их имени происходит греч. Χάλυβας — «сталь». Аристотель описал их способ получения стали: халибы несколько раз промывали речной песок их страны — видимо, таким способом (теперь это называют флотацией) выделяли тяжёлую железосодержащую фракцию породы, добавляли какое-то огнеупорное вещество, и плавили в печах особой конструкции; полученный таким образом металл имел серебристый цвет и был нержавеющим. Из этого процесса, видимо, возникло и название «руда», которое на латыни значит «мокрый» — то есть, «вымытый».
В качестве сырья для выплавки стали использовались магнетитовые пески, которые часто встречаются по всему побережью Чёрного моря: эти магнетитовые пески состоят из смеси мелких зёрен магнетита, титано-магнетита или ильменита, и обломков других пород, так что выплавляемая халибами сталь была легированной, и обладала отличными свойствами. Такой своеобразный способ получения железа не из руды говорит о том, что халибы, в основном, распространили железо как технологический материал, но их способ не мог быть методом повсеместного промышленного производства железных изделий. Однако их производство послужило толчком для дальнейшего развития металлургии железа.
Судя по греческому названию инструментальных металлов χαλκός (это слово обозначает и бронзу, и железо), можно понять, что арийские племена нашли способ выделки железа во время перехода в Азию через Кавказ, а именно — в Колхиде (др.-греч. Κολχίς), так как другого удобного сухопутного пути из Европы в Азию не было. Пройдя степи Причерноморья, они оставили многочисленные памятники культуры бронзового века (так называемая «пахотно-скотоводческая культура»), и двинулись дальше — на юг. Конечно же, по пути они искали сырьё для изготовления бронзовых орудий, и так обнаружили свойства причерноморских песков, дающих новый твёрдый металл — железо. Видимо, сперва они приняли его за олово (первые металлурги плохо различали металлы), и это подтверждается также тем, что название «сталь» в языках северных арийцев (романских, германских, славянских) явно происходит от слова «STANN» через аберрацию N-L, а у римлян это слово обозначало олово. То есть, пытаясь найти олово для бронзы, они обнаружили металл, который оказался крепким и без сплавления с медью, и стали называть его по аналогии с оловом. Найденный тогда способ выплавки стальных изделий не позволял получать их в больших количествах, однако использовался более тысячи лет, пока не была разработана технология выплавки железа из руды, добываемой в копях.
Климент Александрийский в своём энциклопедическом труде «Строматы» упоминает, что по греческим преданиям железо (видимо, выплавка его из руды) было открыто на горе Иде — так называлась горная цепь возле Трои (в Илиаде она упоминается как гора Ида, с которой Зевс наблюдал за битвой греков с троянцами). Произошло это через 73 года после Девкалионова потопа, а этот потоп, согласно Паросской хронике, был в 1528 году до нашей эры, то есть метод выплавки железа из руды был открыт примерно в 1455 году до н. э. Однако из описания Климента не ясно, говорит ли он именно об этой горе в Передней Азии (Ида Фригийская у Вергилия), или же о горе Ида на острове Крит, о которой римский поэт Вергилий в Энеиде пишет:
Остров Юпитера, Крета, лежит средь широкого моря,
Нашего племени там колыбель, где высится Ида …
А римляне, как известно, были потомками малоазиатских троянцев, переселившихся в Италию после разрушения Трои. Могила их предводителя Энея до сих пор существует в местечке Пратика-ди-Маре возле Рима, и в ней был обнаружен железный жезл — символ власти, и другие предметы из железа и бронзы.
Более вероятно, что Климент Александрийский говорит именно о фригийской Иде возле Трои, так как там были найдены древние железные копи и очаги железоделательного производства. Видимо, ознакомившись с методом халибов, древние троянцы развили свой способ выплавки стали из руды, оказавшийся более производительным.
В самой глубокой древности железо ценилось дороже золота, и по описанию Страбона, у африканских племён за 1 фунт железа давали 10 фунтов золота, а по исследованиям историка Г. Арешяна стоимости меди, серебра, золота и железа у древних хеттов были в соотношении 1 : 160 : 1280 : 6400. В те времена железо использовалось как ювелирный металл, из него делали троны и другие регалии царской власти: например, в библейской книге Второзаконие 3,11 описан «одр железный» рефаимского царя Ога. В гробнице Тутанхамона (около 1350 года до н. э.) был найден кинжал из железа в золотой оправе — возможно, подаренный хеттами в дипломатических целях. Но хетты не стремились к широкому распространению железа и его технологий, что видно и из дошедшей до нас переписки египетского фараона и его тестя — царя Хеттов. Фараон просит прислать побольше железа, а царь хеттов уклончиво отвечает, что запасы железа иссякли, а кузнецы заняты на сельскохозяйственных работах, поэтому он не может выполнить просьбу царственного зятя. Как видно, хетты старались использовать свои знания для достижения военных преимуществ, и не давали другим возможности сравняться с ними. Видимо, поэтому железные изделия получили широкое распространение только после Троянской войны и падения державы хеттов, когда благодаря торговой активности греков технология железа стала известной многим, и были открыты железные месторождения и рудники. Так на смену «Бронзовому» веку настал век «Железный».
По описаниям Гомера, хотя во время Троянской войны (примерно 1250 год до н. э.) оружие было в основном из меди и бронзы, но железо уже было хорошо известно и пользовалось большим спросом, хотя больше как драгоценный металл. Например, в 23-й песне «Илиады» Гомер рассказывает, что Ахилл наградил диском из железной крицы победителя в соревновании по метанию диска. Это железо ахейцы добывали у троянцев и сопредельных народов (Илиада 7,473), в том числе у халибов, которые воевали на стороне троянцев:
Прочие мужи ахейские меной вино покупали,
Те за звенящую медь, за седое железо меняли,
Те за воловые кожи или волов круторогих,
Те за своих полоненых. И пир уготовлен веселый…
Возможно, железо было одной из причин, побудивших греков-ахейцев двинуться в Малую Азию, где они узнали секреты его производства. А раскопки в Афинах показали, что уже около 1100 года до н. э. и позднее уже широко были распространены железные мечи, копья, топоры, и даже железные гвозди. В библейской книге Иисуса Навина 17,16 (ср. Судей 14,4) описывается, что филистимляне (библейские «PILISTIM», а это были протогреческие племена, родственные позднейшим эллинам, в основном пеласги) имели множество железных колесниц, то есть, в это время железо уже стало широко применяться в больших количествах.
Гомер в «Илиаде» и «Одиссее» называет железо «многотрудный металл», и описывает закалку орудий:
Расторопный ковач, изготовив топор иль секиру,
В воду металл, раскаливши его, чтоб двойную
Он крепость имел, погружает…
Гомер называет железо многотрудным, потому что в древности основным методом его получения был сыродутный процесс: перемежающиеся слои железной руды и древесного угля прокаливались в специальных печах (горнах — от древнего «Horn» — рог, труба, первоначально это была просто труба, вырытая в земле, обычно горизонтально в склоне оврага). В горне окислы железа восстанавливаются до металла раскалённым углём, который отбирает кислород, окисляясь до окиси углерода, и в результате такого прокаливания руды с углём получалось тестообразное кричное (губчатое) железо. Крицу очищали от шлаков ковкой, выдавливая примеси сильными ударами молота. Первые горны имели сравнительно низкую температуру — заметно меньше температуры плавления чугуна, поэтому железо получалось сравнительно малоуглеродистым. Чтобы получить крепкую сталь приходилось много раз прокаливать и проковывать железную крицу с углём, при этом поверхностный слой металла дополнительно насыщался углеродом и упрочнялся. И хотя это требовало больших трудов, изделия, полученные таким способом, были существенно более крепкими, чем бронзовые.
В дальнейшем научились делать более эффективные печи (в русском языке — домна, домница) для производства стали, и применили меха для подачи воздуха в горн. Уже римляне умели доводить температуру в печи до плавления стали (около 1400 градусов, а чистое железо плавится при 1535 градусах). При этом образуется чугун с температурой плавления 1100—1200 градусов, очень хрупкий в твёрдом состоянии (даже не поддающийся ковке), и не обладающий упругостью стали. Первоначально его считали вредным побочным продуктом (англ. pig iron, по-русски, свинское железо, чушки, откуда, собственно, и происходит слово чугун), но потом обнаружилось, что при повторном прожигании в печи с усиленным продуванием воздуха чугун превращается в сталь хорошего качества, так как лишний углерод выгорает. Такой двухстадийный процесс производства стали из чугуна оказался более простым и выгодным, чем кричный, и этот принцип используется без особых изменений многие века, оставаясь и до наших дней основным способом производства железных материалов.
Происхождение названия

![]()
Схема атома железа (условно)
Версии происхождения славянского слова «железо» (белор. жалеза, болг. желязо, укр. залізо, польск. Żelazo, словен. Železo).
Наиболее вероятно, что это название происходит от древнеарийского корня «ZIL», которым обозначали олово и вообще белые металлы (в том числе серебро — «zilber», и название «цинк» получилось из этого же слова аберрацией L-N). От него же, видимо, происходит и санскритское «жальжа», что означает «металл, руда». Другая версия усматривает в слове славянский корень «лез», тот же, что и в слове «лезвие» (так как железо в основном употреблялось на изготовление оружия), третье связывает с греческим словом χαλκός, что означало железо и медь. Есть также связь между словом «желе» и студнеобразной консистенцией «болотной руды», из которой некоторое время добывался металл.
Название природного карбоната железа (сидерита) происходит от sidereus — звёздный; действительно, первое железо, попавшее в руки людям, было метеоритного происхождения. Возможно, это совпадение не случайно. В частности древнегреческое слово сидерос (σίδηρος) для железа и латинское sidus, означающее «звезда», вероятно, имеют общее происхождение.
Изотопы железа
Изотоп железа 56Fe относится к наиболее стабильным ядрам: все следующие элементы могут уменьшить энергию связи на нуклон путём распада, а все предыдущие элементы, в принципе, могли бы уменьшить энергию связи на нуклон за счёт синтеза. Полагают, что железом оканчивается ряд синтеза элементов в ядрах нормальных звёзд, а все последующие элементы могут образоваться только в результате взрывов сверхновых.
Геохимия железа

![]()
Гидротермальный источник с железистой водой. Окислы железа окрашивают воду в бурый цвет
Железо — один из самых распространённых элементов в Солнечной системе, особенно на планетах земной группы, в частности, на Земле. Значительная часть железа планет земной группы находится в ядрах планет, где его содержание, по оценкам, около 90 %. Содержание железа в земной коре составляет 5 %, а в мантии около 12 %. Из металлов железо уступает по распространённости в коре только алюминию. При этом в ядре находится около 86 % всего железа, а в мантии 14 %.
Геохимические свойства железа
Важнейшая геохимическая особенность железа — наличие у него нескольких степеней окисления. Железо в нейтральной форме — металлическое — слагает ядро земли, возможно, присутствует в мантии и очень редко встречается в земной коре. Закисное железо FeO — основная форма нахождения железа в мантии и земной коре. Окисное железо Fe2O3 характерно для самых верхних, наиболее окисленных, частей земной коры, в частности, осадочных пород.
По кристаллохимическим свойствам ион Fe2+ близок к ионам Mg2+ и Ca2+ — другим главным элементам, составляющим значительную часть всех земных пород. В силу кристаллохимического сходства железо замещает магний и, частично, кальций во многих силикатах. При этом содержание железа в минералах переменного состава обычно увеличивается с уменьшением температуры.
Минералы железа
В земной коре железо распространено достаточно широко — на его долю приходится около 4,1 % массы земной коры (4-е место среди всех элементов, 2-е среди металлов). В мантии и земной коре железо сосредоточено главным образом в силикатах, при этом его содержание значительно в основных и ультраосновных породах, и мало — в кислых и средних породах.
Известно большое число руд и минералов, содержащих железо. Наибольшее практическое значение имеют красный железняк (гематит, Fe2O3; содержит до 70 % Fe), магнитный железняк (магнетит, FeFe2O4, Fe3O4; содержит 72,4 % Fe), бурый железняк или лимонит (гётит и гидрогётит, соответственно FeOOH и FeOOH·nH2O), а также шпатовый железняк (сидерит, карбонат железа(II), FeCO3; содержит около 48 % Fe). Гётит и гидрогётит чаще всего встречаются в корах выветривания, образуя так называемые «железные шляпы», мощность которых достигает несколько сотен метров. Также они могут иметь осадочное происхождение, выпадая из коллоидных растворов в озёрах или прибрежных зонах морей. При этом образуются оолитовые, или бобовые, железные руды. В них часто встречается вивианит Fe(3PO4)2·8H2O, образующий чёрные удлинённые кристаллы и радиально-лучистые агрегаты.
В природе также широко распространены сульфиды железа — пирит FeS2 (серный или железный колчедан) и пирротин. Они не являются железной рудой — пирит используют для получения серной кислоты, а пирротин часто содержит никель и кобальт.
По запасам железных руд Россия занимает первое место в мире. Содержание железа в морской воде — 1×10−5—1×10−8 %.
Получение
В промышленности железо получают из железной руды, в основном из гематита (Fe2O3) и магнетита (Fe3O4).
Существуют различные способы извлечения железа из руд. Наиболее распространённым является доменный процесс.
Первый этап производства — восстановление железа углеродом в доменной печи при температуре 2000 °C. В доменной печи углерод в виде кокса, железная руда в виде агломерата или окатышей и флюс (например, известняк) подаются сверху, а снизу их встречает поток нагнетаемого горячего воздуха.
В печи углерод кокса окисляется до монооксида углерода (угарного газа) кислородом воздуха:
2C + O2 → 2CO↑.
В свою очередь, угарный газ восстанавливает железо из руды:
3CO + Fe2O3 → 2Fe + 3CO2↑.
Флюс добавляется для извлечения нежелательных примесей из руды, в первую очередь силикатов, таких как кварц (диоксид кремния). Типичный флюс содержит известняк (карбонат кальция) и доломит (карбонат магния). Против других примесей используют другие флюсы.
Действие флюса: карбонат кальция под действием тепла разлагается до оксида кальция (негашёная известь):
CaCO3 → CaO + CO2↑.
Оксид кальция соединяется с диоксидом кремния, образуя шлак:
CaO + SiO2 → CaSiO3.
Шлак, в отличие от диоксида кремния, плавится в печи. Более лёгкий, чем железо, шлак плавает на поверхности, и его можно сливать отдельно от металла. Шлак затем употребляется в строительстве и сельском хозяйстве. Расплав железа, полученный в доменной печи, содержит довольно много углерода (чугун). Кроме случаев, когда чугун используется непосредственно, он требует дальнейшей переработки.
Излишний углерод и другие примеси (сера, фосфор) удаляют из чугуна окислением в мартеновских печах или в конвертерах. Электрические печи используют и для выплавки легированных сталей.
Кроме доменного процесса, распространён процесс прямого получения железа. В этом случае предварительно измельчённую руду смешивают с особой глиной, формируя окатыши. Окатыши обжигают, и обрабатывают в шахтной печи горячими продуктами конверсии метана, содержащими водород. Водород легко восстанавливает железо, при этом не происходит загрязнения железа такими примесями как сера и фосфор — обычными примесями в каменном угле. Железо получается в твёрдом виде, и в дальнейшем переплавляется в электрических печах.
Химически чистое железо получается электролизом растворов его солей.
Физические свойства
Железо — типичный металл, в свободном состоянии — серебристо-белого цвета с сероватым оттенком. Чистый металл пластичен, различные примеси (в частности — углерод) повышают его твёрдость и хрупкость. Обладает ярко выраженными магнитными свойствами. Часто выделяют так называемую «триаду железа» — группу трёх металлов (железо Fe, кобальт Co, никель Ni), обладающих схожими физическими свойствами, атомными радиусами и значениями электроотрицательности.
Для железа характерен полиморфизм, он имеет четыре кристаллические модификации:
- до 769 °C существует α-Fe (феррит) с объёмноцентрированной кубической решёткой и свойствами ферромагнетика (769 °C ≈ 1043 K — точка Кюри для железа)
- в температурном интервале 769—917 °C существует β-Fe, который отличается от α-Fe только параметрами объёмноцентрированной кубической решётки и магнитными свойствами парамагнетика
- в температурном интервале 917—1394 °C существует γ-Fe (аустенит) с гранецентрированной кубической решёткой
- выше 1394 °C устойчив δ-Fe с объёмоцентрированной кубической решёткой
Металловедение не выделяет β-Fe как отдельную фазу[4], и рассматривает её как разновидность α-Fe. При нагреве железа или стали выше точки Кюри (769 °C ≈ 1043 K) тепловое движение ионов расстраивает ориентацию спиновых магнитных моментов электронов, ферромагнетик становится парамагнетиком — происходит фазовый переход второго рода, но фазового перехода первого рода с изменением основных физических параметров кристаллов не происходит.
Для чистого железа при нормальном давлении, с точки зрения металловедения, существуют следующие устойчивые модификации:
- От абсолютного нуля до 910 °C устойчива α-модификация с объёмноцентрированной кубической (ОЦК) кристаллической решёткой. Твёрдый раствор углерода в α-железе называется ферритом.
- От 910 до 1400 °C устойчива γ-модификация с гранецентрированной кубической (ГЦК) кристаллической решёткой. Твёрдый раствор углерода в γ-железе называется аустенитом.
- От 910 до 1539 °C устойчива δ-модификация с объёмноцентрированной кубической (ОЦК) кристаллической решёткой. Твёрдый раствор углерода в δ-железе (также как и в α-железе) называется ферритом. Иногда различают высокотемпературный δ-феррит и низкотемпературный α-феррит (или просто феррит), хотя их атомные структуры одинаковы.
Наличие в стали углерода и легирующих элементов существенным образом изменяет температуры фазовых переходов (см. фазовую диаграмму железо — углерод).
- В области высоких давлений (свыше 104 МПа, 100 тыс. атм.) возникает модификация ε-железа с гексагональной плотноупакованной (ГПУ) решёткой.
Явление полиморфизма чрезвычайно важно для металлургии стали. Именно благодаря α—γ переходам кристаллической решётки происходит термообработка стали. Без этого явления железо как основа стали не получило бы такого широкого применения.
Железо тугоплавко, относится к металлам средней активности. Температура плавления железа 1539 °C, температура кипения — 2862 °C.
Химические свойства
Основные степени окисления железа — +2 и +3.
При хранении на воздухе при температуре до 200 °C железо постепенно покрывается плотной плёнкой оксида, препятствующего дальнейшему окислению металла. Во влажном воздухе железо покрывается рыхлым слоем ржавчины, который не препятствует доступу кислорода и влаги к металлу и его разрушению. Ржавчина не имеет постоянного химического состава, приближённо её химическую формулу можно записать как Fe2O3·xH2O.
С кислородом железо реагирует при нагревании. При сгорании железа на воздухе образуется оксид Fe3O4, при сгорании в чистом кислороде — оксид Fe2O3. Если кислород или воздух пропускать через расплавленное железо, то образуется оксид FeO. При нагревании порошка серы и железа образуется сульфид, приближённую формулу которого можно записать как FeS.
Железо при нагревании реагирует с галогенами. Так как FeF3 нелетуч, железо устойчиво к действию фтора до температуры 200—300 °C. При хлорировании железа (при температуре около 200 °C) образуется летучий FeCl3. Если взаимодействие железа и брома протекает при комнатной температуре или при нагревании и повышенном давлении паров брома, то образуется FeBr3. При нагревании FeCl3 и, особенно, FeBr3 отщепляют галоген и превращаются в галогениды железа(II). При взаимодействии железа и иода образуется иодид Fe3I8.
При нагревании железо реагирует с азотом, образуя нитрид железа Fe3N, с фосфором, образуя фосфиды FeP, Fe2P и Fe3P, с углеродом, образуя карбид Fe3C, с кремнием, образуя несколько силицидов, например, FeSi.
При повышенном давлении металлическое железо реагирует с оксидом углерода(II) CO, причём образуется жидкий, при обычных условиях легко летучий пентакарбонил железа Fe(CO)5. Известны также карбонилы железа составов Fe2(CO)9 и Fe3(CO)12. Карбонилы железа служат исходными веществами при синтезе железоорганических соединений, в том числе и ферроцена состава (η5-C5H5)2Fe.
Чистое металлическое железо устойчиво в воде и в разбавленных растворах щелочей. В концентрированной серной и азотной кислотах железо не растворяется, так как прочная оксидная плёнка пассивирует его поверхность.
С соляной и разбавленной (приблизительно 20%-й) серной кислотами железо реагирует с образованием солей железа(II):
Fe + 2HCl → FeCl2 + H2↑;
Fe + H2SO4 → FeSO4 + H2↑.
При взаимодействии железа с приблизительно 70%-й серной кислотой реакция протекает с образованием сульфата железа(III):
2Fe + 6H2SO4 → Fe2(SO4)3 + 3SO2↑ + 6H2O.
Оксид железа(II) FeO обладает основными свойствами, ему отвечает основание Fe(OH)2. Оксид железа(III) Fe2O3 слабо амфотерен, ему отвечает ещё более слабое, чем Fe(OH)2, основание Fe(OH)3, которое реагирует с кислотами:
2Fe(OH)3 + 3H2SO4 → Fe2(SO4)3 + 6H2O.
Гидроксид железа(III) Fe(OH)3 проявляет слабо амфотерные свойства, он способен реагировать только с концентрированными растворами щелочей:
Fe(OH)3 + 3КОН → K3[Fe(OH)6].
Образующиеся при этом гидроксокомплексы железа(III) устойчивы в сильно щелочных растворах. При разбавлении растворов водой они разрушаются, причём в осадок выпадает Fe(OH)3.
Соединения железа(III) в растворах восстанавливаются металлическим железом:
Fe + 2FeCl3 → 3FeCl2.
При хранении водных растворов солей железа(II) наблюдается окисление железа(II) до железа(III):
4FeCl2 + O2 + 2H2O → 4Fe(OH)Cl2.
Из солей железа(II) в водных растворах устойчива соль Мора — двойной сульфат аммония и железа(II) (NH4)2Fe(SO4)2·6Н2O.
Железо(III) способно образовывать двойные сульфаты с однозарядными катионами типа квасцов, например, KFe(SO4)2 — железокалиевые квасцы, (NH4)Fe(SO4)2 — железоаммонийные квасцы и т. д.
При действии газообразного хлора или озона на щелочные растворы соединений железа(III) образуются соединения железа(VI) — ферраты, например, феррат(VI) калия K2FeO4. Имеются сообщения о получении под действием сильных окислителей соединений железа(VIII).
Для обнаружения в растворе соединений железа(III) используют качественную реакцию ионов Fe3+ с тиоцианат-ионами SCN—. При взаимодействии ионов Fe3+ с анионами SCN— образуется ярко-красный роданид железа Fe(SCN)3. Другим реактивом на ионы Fe3+ служит гексацианоферрат(II) калия K4[Fe(CN)6] (жёлтой кровяная соль). При взаимодействии ионов Fe3+ и [Fe(CN)6]4− выпадает ярко-синий осадок берлинской лазури:
4K4[Fe(CN)6] + 4Fe3+ → 4KFeIII[FeII(CN)6]↓ + 12K+.
Реактивом на ионы Fe2+ в растворе может служить гексацианоферрат(III) калия K3[Fe(CN)6] (красная кровяная соль). При взаимодействии ионов Fe2+ и [Fe(CN)6]3− выпадает осадок турнбулевой сини:
3K3[Fe(CN)6] + 3Fe2+ → 3KFeII[FeIII(CN)6]↓ + 6K+.
Интересно, что берлинская лазурь и турнбулева синь — две формы одного и того же вещества, так как в растворе устанавливается равновесие:
KFeIII[FeII(CN)6] ↔ KFeII[FeIII(CN)6].
Применение

![]()
Железная руда
Железо — один из самых используемых металлов, на него приходится до 95 % мирового металлургического производства.
-
Железо является основным компонентом сталей и чугунов — важнейших конструкционных материалов.
Железо может входить в состав сплавов на основе других металлов — например, никелевых.
Магнитная окись железа (магнетит) — важный материал в производстве устройств долговременной компьютерной памяти: жёстких дисков, дискет и т. п.
Ультрадисперсный порошок магнетита используется в черно-белых лазерных принтерах в качестве тонера.
Уникальные ферромагнитные свойства ряда сплавов на основе железа способствуют их широкому применению в электротехнике для магнитопроводов трансформаторов и электродвигателей.
Хлорид железа(III) (хлорное железо) используется в радиолюбительской практике для травления печатных плат.
Семиводный сульфат железа (железный купорос) в смеси с медным купоросом используется для борьбы с вредными грибками в садоводстве и строительстве.
Железо применяется в качестве анода в железо-никелевых аккумуляторах, железо-воздушных аккумуляторах.
Биологическое значение железа
В живых организмах железо является важным микроэлементом, катализирующим процессы обмена кислородом (дыхания). В организме взрослого человека содержится около 3,5 грамма железа (около 0,02 %), из которых 75 % являются главным действующим элементом гемоглобина крови, остальное входит в состав ферментов других клеток, катализируя процессы дыхания в клетках. Недостаток железа проявляется как болезнь организма (хлороз у растений и анемия у животных).
Обычно железо входит в ферменты в виде комплекса, называемого гемом. В частности, этот комплекс присутствует в гемоглобине — важнейшем белке, обеспечивающем транспорт кислорода с кровью ко всем органам человека и животных. И именно он окрашивает кровь в характерный красный цвет.
Комплексы железа, отличные от гема, встречаются, например, в ферменте метан-моноксигеназе, окисляющем метан в метанол, в важном ферменте рибонуклеотид-редуктазе, который участвует в синтезе ДНК.
Неорганические соединения железа встречается в некоторых бактериях, иногда используется ими для связывания азота воздуха.
В организм животных и человека железо поступает с пищей (наиболее богаты им печень, мясо, яйца, бобовые, хлеб, крупы, свёкла). Интересно, что некогда шпинат ошибочно был внесён в этот список (из-за опечатки в результатах анализа — был потерян «лишний» ноль после запятой).
Суточная потребность человека в железе следующая: дети — от 4 до 18 мг, взрослые мужчины — 10 мг, взрослые женщины — 18 мг, беременные женщины во второй половине беременности — 33 мг. У женщин потребность несколько выше, чем у мужчин. Как правило, железа, поступающего с пищей, вполне достаточно, но в некоторых специальных случаях (анемия, а также при донорстве крови) необходимо применять железосодержащие препараты и пищевые добавки (гематоген, ферроплекс).
Содержание железа в воде больше 1—2 мг/л значительно ухудшает её органолептические свойства, придавая ей неприятный вяжущий вкус, и делает воду малопригодной для использования, вызывает у человека аллергические реакции, может стать причиной болезни крови и печени (гемохроматоз). ПДК железа в воде 0,3 мг/л.
Избыточная доза железа (200 мг и выше) может оказывать токсическое действие. Передозировка железа угнетает антиоксидантную систему организма, поэтому употреблять препараты железа здоровым людям не рекомендуется.
Соединения железа
Оксиды железа
Гидроксиды железа
Железнение
Железо самородное
III. Роль железа в жизни человека и растений
Биохимики открыли важную роль железа в жизни растений, животных и человека. Входя в состав чрезвычайно сложно построенного органического соединения, называемого гемоглобином, железо обусловливает красную окраску этого вещества, от которого в свою очередь, зависит цвет крови человека и животных. В организме взрослого человека содержится 3 г чистого железа, 75% которого входит в состав гемоглобина. Основная роль гемоглобина – перенос кислорода из легких к тканям, а в обратном направлении – CO2.
Железо необходимо и растениям. Оно входит в состав цитоплазмы, участвует в процессе фотосинтеза. Растения, выращенные на субстрате, не содержащем железа, имеют белые листья. Маленькая добавка железа к субстрату – и они приобретают зеленый цвет. Больше того, стоит белый лист смазать раствором соли, содержащей железо, и вскоре смазанное место зеленеет.
Так от одной и той же причины – наличия железа в соках и тканях – весело зеленеют листья растений и ярко румянятся щеки человека.
IV. Физические свойства железа
Железо – это серебристо-белый металл с температурой плавления 1539оС. Очень пластичный, поэтому легко обрабатывается, куется, прокатывается, штампуется. Железо обладает способностью намагничиваться и размагничиваться, поэтому применяется в качестве сердечников электромагнитов в различных электрических машинах и аппаратах. Ему можно придать большую прочность и твердость методами термического и механического воздействия, например, с помощью закалки и прокатки.
Различают химически чистое и технически чистое железо. Технически чистое железо, по сути, представляет собой низкоуглеродистую сталь, оно содержит 0,02-0,04% углерода, а кислорода, серы, азота и фосфора – еще меньше. Химически чистое железо содержит менее 0,01% примесей. Химически чистое железо – серебристо-серый, блестящий, по внешнему виду очень похожий на платину металл. Химически чистое железо устойчиво к коррозии и хорошо сопротивляется действию кислот. Однако ничтожные доли примесей лишают его этих драгоценный свойств.
V. Получение железа
Восстановлением из оксидов углём или оксидом углерода (II), а также водородом:
FeO + C = Fe + CO
Fe2O3 + 3CO = 2Fe + 3CO2
Fe2O3 + 3H2 = 2Fe + 3H2O
Опыт «Получение железа алюминотермией»
VI. Химические свойства железа
Как элемент побочной подгруппы железо может проявлять несколько степеней окисления. Мы рассмотрим только соединения, в которых железо проявляет степени окисления +2 и +3. Таким образом, можно говорить, что у железа имеется два ряда соединений, в которых оно двух- и трехвалентно.
1) На воздухе железо легко окисляется в присутствии влаги (ржавление):
4Fe + 3O2 + 6H2 O = 4Fe(OH)3
2) Накалённая железная проволока горит в кислороде, образуя окалину — оксид железа (II, III) — вещество чёрного цвета:
3Fe + 2O2 = Fe3O4
При пропускании кислорода через расплавленное железо возможно образование оксида железа (II):
2Fe+O2=2FeO
C кислородом во влажном воздухе образуется Fe2O3‧nH2O
Опыт «Горение железа в кислороде«
3) При высокой температуре (700–900°C) железо реагирует с парами воды:
3Fe + 4H2O =t˚C= Fe3O4 + 4H2
4) Железо реагирует с неметаллами при нагревании:
Железо реагирует с галогенами с образованием галогенидов. При этом активные неметаллы (фтор, хлор и бром) окисляют железо до степени окисления +3:
2Fe + 3Cl2 =t= 2FeCl3
Менее активный йод окисляет железо до степени окисления +2:
Fe + I2 =t= FeI2
Железо реагирует с серой с образованием сульфида железа (II):
Fe + S =t= FeS
Железо реагирует с фосфором. При этом образуется бинарное соединения – фосфид железа:
Fe + P =t= FeP
С азотом железо реагирует при нагревании с образованием нитрида:
6Fe + N2 =t= 2Fe3N
Железо реагирует с углеродом и кремнием с образованием карбида и силицида:
3Fe + C =t= Fe3C
5) Железо легко растворяется в соляной и разбавленной серной кислотах при обычных условиях:
Fe + 2HCl = FeCl2 + H2
Fe + H2SO4(разб.) = FeSO4 + H2
6) В концентрированных кислотах – окислителях железо растворяется только при нагревании
При обычных условиях железо не реагирует с концентрированной серной кислотой из-за пассивации – образования плотной оксидной пленки. При нагревании реакция идет, образуются оксид серы (IV), сульфат железа (III) и вода:
2Fe + 6H2SO4(конц.) =t= Fe2(SO4)3 + 3SO2 + 6H2O
Железо не реагирует при обычных условиях с концентрированной азотной кислотой также из-за пассивации. При нагревании реакция идет с образованием нитрата железа (III), оксида азота (IV) и воды:
Fe+6HNO3(конц.) =t= Fe(NO3)3+3NO2+3H2O
С разбавленной азотной кислотой железо реагирует с образованием оксида азота (II):
Fe+4HNO3(разб.гор.) =t= Fe(NO3)3+NO+2H2O
При взаимодействии железа с очень разбавленной азотной кислотой образуется нитрат аммония:
8Fe+30HNO3(оч. разб.) =t= 8Fe(NO3)3+3NH4NO3+9H2O
Опыт «Взаимодействие железа с концентрированными кислотами»
7) Железо вытесняет металлы, стоящие правее его в ряду напряжений из растворов их солей.
Fe + CuSO4 = FeSO4 + Cu
 Железо может реагировать с щелочными растворами или расплавами сильных окислителей. При этом железо окисляет до степени окисления +6, образуя соль (феррат)
Железо может реагировать с щелочными растворами или расплавами сильных окислителей. При этом железо окисляет до степени окисления +6, образуя соль (феррат)
При взаимодействии железа с расплавом нитрата калия в присутствии гидроксида калия железо окисляется до феррата калия, а азот восстанавливается либо до нитрита калия, либо до аммиака:
Fe+2KOH+3KNO3=3KNO2+K2FeO4+H2O
9) Простое вещество железо восстанавливает железо до степени окисления +2 при взаимодействии с соединениями железа +3:
2Fe(NO3)3+Fe=3Fe(NO3)2
2FeCl3+Fe=3FeCl2
Fe2(SO4)3+Fe=3FeSO4
VII. Качественные реакции на
Железо (II)
Железо (III)
VIII. Применение железа
Основная часть получаемого в мире железа используется для получения чугуна и стали — сплавов железа с углеродом и другими металлами. Чугуны содержат около 4% углерода. Стали содержат углерода менее 1,4%.
Чугуны необходимы для производства различных отливок — станин тяжелых машин и т.п.
Изделия из чугуна
Стали используются для изготовления машин, различных строительных материалов, балок, листов, проката, рельсов, инструмента и множества других изделий. Для производства различных сортов сталей применяют так называемые легирующие добавки, которыми служат различные металлы: Мn, Сr, Мо и другие, улучшающие качество стали.
Изделия из стали
«Появление железа»
Это интересно
Железо, свойства атома, химические и физические свойства.
Fe 26 Железо
55,845(2) 1s2 2s2 2p6 3s2 3p6 3d6 4s2
Железо — элемент периодической системы химических элементов Д. И. Менделеева с атомным номером 26. Расположен в 8-й группе (по старой классификации — побочной подгруппе восьмой группы), четвертом периоде периодической системы.
Общие сведения
Свойства атома железа
Химические свойства железа
Физические свойства железа
Кристаллическая решётка железа
Дополнительные сведения
Таблица химических элементов Д.И. Менделеева
Общие сведения:
| 100 | Общие сведения | |
| 101 | Название | Железо |
| 102 | Прежнее название | |
| 103 | Латинское название | Ferrum |
| 104 | Английское название | Iron |
| 105 | Символ | Fe |
| 106 | Атомный номер (номер в таблице) | 26 |
| 107 | Тип | Металл |
| 108 | Группа | Амфотерный, переходный, чёрный металл |
| 109 | Открыт | Известно с глубокой древности |
| 110 | Год открытия | до 5000 года до н.э. |
| 111 | Внешний вид и пр. | Ковкий, вязкий металл серебристо-белого цвета с сероватым оттенком |
| 112 | Происхождение | Природный материал |
| 113 | Модификации | |
| 114 | Аллотропные модификации | 5 аллотропных модификаций железа: — α-железо (феррит) с кубической объемно-центрированной кристаллической решёткой и свойствами ферромагнетика, — β-железо с кубической объёмно-центрированной кристаллической решёткой, отличающееся от α-железа параметрами кристаллической решётки и свойствами парамагнетика. β-железо служит для обозначения α-железа выше точки Кюри (точка Кюри железа 769 °C), — γ-железо (аустенит) с кубической гранецентрированной кристаллической решёткой, — δ-железо с кубической объёмно-центрированной кристаллической решёткой, — ε-железо с гексагональной плотноупакованной кристаллической решёткой |
| 115 | Температура и иные условия перехода аллотропных модификаций друг в друга* | — α-железо (феррит) существует при температуре ниже 769 °C и иных стандартных условиях (точка Кюри железа 769 °C), — β-железо существует в интервале температур от 769 °C до 917 °C и иных стандартных условиях, — γ-железо (аустенит) существует в интервале температур от 917 °C до 1394 °C и иных стандартных условиях, — δ-железо существует при температуре выше 1394 °C и иных стандартных условиях, — ε-железо существует при давлении более 13 ГПа и иных стандартных условиях |
| 116 | Конденсат Бозе-Эйнштейна | |
| 117 | Двумерные материалы | |
| 118 | Содержание в атмосфере и воздухе (по массе) | 0 % |
| 119 | Содержание в земной коре (по массе) | 6,3 % |
| 120 | Содержание в морях и океанах (по массе) | 3,0·10-7 % |
| 121 | Содержание во Вселенной и космосе (по массе) | 0,11 % |
| 122 | Содержание в Солнце (по массе) | 0,1 % |
| 123 | Содержание в метеоритах (по массе) | 0,22 % |
| 124 | Содержание в организме человека (по массе) | 0,006 % |
Свойства атома железа:
| 200 | Свойства атома | |
| 201 | Атомная масса (молярная масса) | 55,845(2) а.е.м. (г/моль) |
| 202 | Электронная конфигурация | 1s2 2s2 2p6 3s2 3p6 3d6 4s2 |
| 203 | Электронная оболочка | K2 L8 M14 N2 O0 P0 Q0 R0
|
| 204 | Радиус атома (вычисленный) | 156 пм |
| 205 | Эмпирический радиус атома* | 140 пм |
| 206 | Ковалентный радиус* | 123 пм – low-spin, 152 пм – high-spin |
| 207 | Радиус иона (кристаллический) | Fe2+ low spin 75 (6) пм, Fe3+ low spin 69 (6) пм, Fe4+ low spin 72,5 (6) пм, Fe6+ low spin 39 (4) пм, Fe2+ high spin 92 (6) пм, Fe3+ high spin 78,5 (6) пм (в скобках указано координационное число – характеристика, которая определяет число ближайших частиц (ионов или атомов) в молекуле или кристалле) |
| 208 | Радиус Ван-дер-Ваальса | |
| 209 | Электроны, Протоны, Нейтроны | 26 электронов, 26 протонов, 30 нейтронов |
| 210 | Семейство (блок) | элемент d-семейства |
| 211 | Период в периодической таблице | 4 |
| 212 | Группа в периодической таблице | 8-ая группа (по старой классификации – побочная подгруппа 8-ой группы) |
| 213 | Эмиссионный спектр излучения |  |

Химические свойства железа:
| 300 | Химические свойства | |
| 301 | Степени окисления | -4, -2, -1, 0, +1, +2 , +3 , +4, +5, +6 , +7 |
| 302 | Валентность | II, III |
| 303 | Электроотрицательность | 1,83 (шкала Полинга) |
| 304 | Энергия ионизации (первый электрон) | 762,47 кДж/моль (7,9024681(12) эВ) |
| 305 | Электродный потенциал | Fe2+ + 2e— → Fe, Eo = -0,440 В, Fe3+ + e— → Fe2+, Eo = +0,771, Fe3+ + 3e— → Fe, Eo = -0,037 В |
| 306 | Энергия сродства атома к электрону | 14,785(4) кДж/моль (0,153236(35) эВ) |
Физические свойства железа:
| 400 | Физические свойства | |
| 401 | Плотность | 7,874 г/см3 (при 0 °C/20 °C и иных стандартных условиях, состояние вещества – твердое тело), 6,98 г/см3 (при температуре плавления 1538 °C и иных стандартных условиях, состояние вещества – жидкость), 6,9 г/см3 (при 1589 °C и иных стандартных условиях, состояние вещества – жидкость) |
| 402 | Температура плавления* | 1538 °C (1811 K, 2800 °F) |
| 403 | Температура кипения* | 2861 °C (3134 K, 5182 °F) |
| 404 | Температура сублимации | |
| 405 | Температура разложения | |
| 406 | Температура самовоспламенения смеси газа с воздухом | |
| 407 | Удельная теплота плавления (энтальпия плавления ΔHпл)* | 13,81 кДж/моль |
| 408 | Удельная теплота испарения (энтальпия кипения ΔHкип)* | 340 кДж/моль |
| 409 | Удельная теплоемкость при постоянном давлении | 0,448 Дж/г·K (при 25 °C), 0,64 Дж/г·K (при 0-1000 °C) |
| 410 | Молярная теплоёмкость* | 25,10 Дж/(K·моль) |
| 411 | Молярный объём | 7,0923 см³/моль |
| 412 | Теплопроводность | 80,4 Вт/(м·К) (при стандартных условиях), 80,4 Вт/(м·К) (при 300 K) |
| 413 | Коэффициент теплового расширения | 11,8 мкм/(М·К) (при 25 °С) |
| 414 | Коэффициент температуропроводности | |
| 415 | Критическая температура | |
| 416 | Критическое давление | |
| 417 | Критическая плотность | |
| 418 | Тройная точка | |
| 419 | Давление паров (мм.рт.ст.) | 0,01 мм.рт.ст. (при 1425 °C), 0,1 мм.рт.ст. (при 1586 °C), 1 мм.рт.ст. (при 1790 °C), 10 мм.рт.ст. (при 2045 °C), 100 мм.рт.ст. (при 2376 °C) |
| 420 | Давление паров (Па) | 1 Па (при 1728 K), 10 Па (при 1890 K), 100 Па (при 2091 K), 1 кПа (при 2346 K), 10 кПа (при 2679 K), 100 кПа (при 3132 K) |
| 421 | Стандартная энтальпия образования ΔH | 0 кДж/моль (при 298 К, для состояния вещества – твердое тело) |
| 422 | Стандартная энергия Гиббса образования ΔG | 0 кДж/моль (при 298 К, для состояния вещества – твердое тело) |
| 423 | Стандартная энтропия вещества S | 27,15 Дж/(моль·K) (при 298 К, для состояния вещества – твердое тело) |
| 424 | Стандартная мольная теплоемкость Cp | 25 Дж/(моль·K) (при 298 К, для состояния вещества – твердое тело) |
| 425 | Энтальпия диссоциации ΔHдисс | |
| 426 | Диэлектрическая проницаемость | |
| 427 | Магнитный тип | Ферромагнитный материал (ниже 769 °C), парамагнитный материал (выше 769 °C) |
| 428 | Точка Кюри* | 769 °C (1042,15 К, 1416,2 °F) |
| 429 | Объемная магнитная восприимчивость | |
| 430 | Удельная магнитная восприимчивость | |
| 431 | Молярная магнитная восприимчивость | |
| 432 | Электрический тип | Проводник |
| 433 | Электропроводность в твердой фазе | 10,4·106 См/м (при 20 °C) |
| 434 | Удельное электрическое сопротивление | 96,1 нОм·м (при 20 °C) |
| 435 | Сверхпроводимость при температуре | |
| 436 | Критическое магнитное поле разрушения сверхпроводимости | |
| 437 | Запрещенная зона | |
| 438 | Концентрация носителей заряда | |
| 439 | Твёрдость по Моосу | 4,0 |
| 440 | Твёрдость по Бринеллю | 200-1180 МПа |
| 441 | Твёрдость по Виккерсу | 608 МПа |
| 442 | Скорость звука | 5120 м/с (при 20 °C) (тонкий стержень) |
| 443 | Поверхностное натяжение | |
| 444 | Динамическая вязкость газов и жидкостей | |
| 445 | Взрывоопасные концентрации смеси газа с воздухом, % объёмных | |
| 446 | Взрывоопасные концентрации смеси газа с кислородом, % объёмных | |
| 446 | Предел прочности на растяжение | |
| 447 | Предел текучести | |
| 448 | Предел удлинения | |
| 449 | Модуль Юнга | 211 ГПа |
| 450 | Модуль сдвига | 82 ГПа |
| 451 | Объемный модуль упругости | 170 ГПа |
| 452 | Коэффициент Пуассона | 0,29 |
| 453 | Коэффициент преломления |
Кристаллическая решётка железа:
| 500 | Кристаллическая решётка | |
| 511 | Кристаллическая решётка #1 | α-железо (феррит) |
| 512 | Структура решётки | Кубическая объёмно-центрированная
|
| 513 | Параметры решётки | 2,866 Å |
| 514 | Отношение c/a | |
| 515 | Температура Дебая | 460 K |
| 516 | Название пространственной группы симметрии | Im_ 3m |
| 517 | Номер пространственной группы симметрии | 229 |
| 521 | Кристаллическая решётка #2 | γ-железо (аустенит) |
| 522 | Структура решётки | Кубическая гранецентрированная
|
| 523 | Параметры решётки | 3,656 Å |
| 524 | Отношение c/a | |
| 525 | Температура Дебая | |
| 526 | Название пространственной группы симметрии | Fm_ 3m |
| 527 | Номер пространственной группы симметрии | 225 |
| 531 | Кристаллическая решётка #3 | δ-железо |
| 532 | Структура решётки | Кубическая объёмно-центрированная
|
| 533 | Параметры решётки | 2,93 Å |
| 534 | Отношение c/a | |
| 535 | Температура Дебая | |
| 536 | Название пространственной группы симметрии | Im_ 3m |
| 537 | Номер пространственной группы симметрии | 229 |


Дополнительные сведения:
| 900 | Дополнительные сведения | |
| 901 | Номер CAS | 7439-89-6 |
Примечание:
115* Температура и иные условия перехода аллотропных модификаций железа друг в друга согласно [1]:
— α-железо (феррит) существует при температуре ниже 770 °C и иных стандартных условиях (точка Кюри железа согласно [1] 770 °C),
— β-железо существует в интервале температур от 770 °C до 912 °C и иных стандартных условиях,
— γ-железо (аустенит) существует в интервале температур от 912 °C до 1394 °C и иных стандартных условиях,
— δ-железо существует при температуре выше 1394 °C и иных стандартных условиях,
— ε-железо существует при температуре несколько сотен °C и давлении более 10 ГПа либо при более высоком давлении и иных стандартных условиях.
205* Эмпирический радиус атома железа согласно [1] и [3] составляет 126 пм.
206* Ковалентный радиус железа согласно [1] составляет 132±3 пм (low-spin) и 152±6 пм (high-spin), ковалентный радиус железа согласно [3] [Россия] составляет 117 пм.
402* Температура плавления железа согласно [3] и [4] составляет 1538,85 °C (1812 К, 2801,93 °F) и 1539 °C (1812,15 К, 2802,2 °F) соответственно.
403* Температура кипения железа согласно [4] составляет 2870 °C (3143,15 К, 5198 °F).
407* Удельная теплота плавления (энтальпия плавления ΔHпл) железа согласно [3] и [4] составляет 13,8 кДж/моль.
408* Удельная теплота испарения (энтальпия кипения ΔHкип) железа согласно [4] составляет 350 кДж/моль.
410* Молярная теплоемкость железа согласно [3] составляет 25,14 Дж/(K·моль).
428* Точка Кюри железа согласно [1] составляет 770 °C (1043 K, 1418 °F).
Источники:
- https://en.wikipedia.org/wiki/Iron
- https://de.wikipedia.org
- https://ru.wikipedia.org/wiki/Железо
- http://chemister.ru/Database/properties.php?dbid=1&id=236
[know]
Таблица химических элементов Д.И. Менделеева
Коэффициент востребованности
3 159