The Enzyme Commission number (EC number) is a numerical classification scheme for enzymes, based on the chemical reactions they catalyze.[1] As a system of enzyme nomenclature, every EC number is associated with a recommended name for the corresponding enzyme-catalyzed reaction.
EC numbers do not specify enzymes but enzyme-catalyzed reactions. If different enzymes (for instance from different organisms) catalyze the same reaction, then they receive the same EC number.[2] Furthermore, through convergent evolution, completely different protein folds can catalyze an identical reaction (these are sometimes called non-homologous isofunctional enzymes)[3] and therefore would be assigned the same EC number. By contrast, UniProt identifiers uniquely specify a protein by its amino acid sequence.[4]
Format of number[edit]
Every enzyme code consists of the letters «EC» followed by four numbers separated by periods. Those numbers represent a progressively finer classification of the enzyme. Preliminary EC numbers exist and have an ‘n’ as part of the fourth (serial) digit (e.g. EC 3.5.1.n3).[2]
For example, the tripeptide aminopeptidases have the code «EC 3.4.11.4», whose components indicate the following groups of enzymes:
- EC 3 enzymes are hydrolases enzymes (enzymes that use water to break up some other molecule)
- EC 3.4 are hydrolases that act on peptide bonds
- EC 3.4.11 are those hydrolases that cleave off the amino-terminal amino acid from a polypeptide
- EC 3.4.11.4 are those that cleave off the amino-terminal end from a tripeptide
Top level codes[edit]
Top-level EC numbers[5]| Class | Reaction catalyzed | Typical reaction | Enzyme example(s) with trivial name |
|---|---|---|---|
| EC 1 Oxidoreductases | Oxidation/reduction reactions; transfer of H and O atoms or electrons from one substance to another | AH + B → A + BH (reduced) A + O → AO (oxidized) | Dehydrogenase, oxidase |
| EC 2 Transferases | Transfer of a functional group from one substance to another. The group may be methyl-, acyl-, amino- or phosphate group | AB + C → A + BC | Transaminase, kinase |
| EC 3 Hydrolases | Formation of two products from a substrate by hydrolysis | AB + H2O → AOH + BH | Lipase, amylase, peptidase, phosphatase |
| EC 4 Lyases | Non-hydrolytic addition or removal of groups from substrates. C-C, C-N, C-O or C-S bonds may be cleaved | RCOCOOH → RCOH + CO2 or [X-A+B-Y] → [A=B + X-Y] | Decarboxylase |
| EC 5 Isomerases | Intramolecule rearrangement, i.e. isomerization changes within a single molecule | ABC → BCA | Isomerase, mutase |
| EC 6 Ligases | Join together two molecules by synthesis of new C-O, C-S, C-N or C-C bonds with simultaneous breakdown of ATP | X + Y + ATP → XY + ADP + Pi | Synthetase |
| EC 7 Translocases | Catalyse the movement of ions or molecules across membranes or their separation within membranes | Transporter |
Reaction similarity[edit]
Similarity between enzymatic reactions can be calculated by using bond changes, reaction centres or substructure metrics (formerly EC-BLAST], now the EMBL-EBI Enzyme Portal).[6]
History[edit]
Before the development of the EC number system, enzymes were named in an arbitrary fashion, and names like old yellow enzyme and malic enzyme that give little or no clue as to what reaction was catalyzed were in common use. Most of these names have fallen into disuse, though a few, especially proteolyic enzymes with very low specificity, such as pepsin and papain, are still used, as rational classification on the basis of specificity has been very difficult.
By the 1950s the chaos was becoming intolerable, and after Hoffman-Ostenhof[7] and Dixon and Webb[8] had proposed somewhat similar schemes for classifying enzyme-catalyzed reactions, the International Congress of Biochemistry in Brussels set up the Commission on Enzymes under the chairmanship of Malcolm Dixon in 1955. The first version was published in 1961, and the Enzyme Commission was dissolved at that time, though its name lives on in the term EC Number. The current sixth edition, published by the International Union of Biochemistry and Molecular Biology in 1992 as the last version published as a printed book, contains 3196 different enzymes. Supplements 1-4 were published 1993-1999. Subsequent supplements have been published electronically, at the website of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology.[5] In August 2018, the IUBMB modified the system by adding the top-level EC 7 category containing translocases.[9]
See also[edit]
- List of EC numbers
- List of enzymes
- TC number (classification of membrane transport proteins)
References[edit]
- ^ Webb, E. C. (1992). Enzyme nomenclature 1992: recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the nomenclature and classification of enzymes. Academic Press. ISBN 978-0-12-227164-9.
- ^ a b «ENZYME (Enzyme nomenclature database)». ExPASy. Archived from the original on 21 March 2019. Retrieved 24 April 2019.
- ^ Omelchenko MV, Galperin MY, Wolf YI, Koonin EV (2010). «Non-homologous isofunctional enzymes: a systematic analysis of alternative solutions in enzyme evolution». Biology Direct. 5 (1): 31. doi:10.1186/1745-6150-5-31. PMC 2876114. PMID 20433725.
- ^ Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LS (Jan 2004). «UniProt: the Universal Protein knowledgebase». Nucleic Acids Research. 32 (Database issue): D115–9. doi:10.1093/nar/gkh131. PMC 308865. PMID 14681372.
- ^ a b Moss GP. «Recommendations of the Nomenclature Committee». International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes by the Reactions they Catalyse. Archived from the original on 2018-09-10. Retrieved 2006-03-14.
- ^ Rahman SA, Cuesta SM, Furnham N, Holliday GL, Thornton JM (Feb 2014). «EC-BLAST: a tool to automatically search and compare enzyme reactions». Nature Methods. 11 (2): 171–174. doi:10.1038/nmeth.2803. PMC 4122987. PMID 24412978.
- ^ Hoffman-Ostenhof, O (1953). «Suggestions for a more rational classification and nomenclature of enzymes». Advances in Enzymology and Related Areas of Molecular Biology. Advances in Enzymology and Related Subjects of Biochemistry. Vol. 14. pp. 219–260. doi:10.1002/9780470122594.ch7. ISBN 9780470122594. PMID 13057718.
- ^ Dixon, M; Webb, E.C. (1958). Enzymes. London: Longmans Green. pp. 183–227.
- ^ Tipton, Keith (August 2018). «Enzyme Nomenclature News: Translocases (EC 7): A new EC Class». ExplorEnz: the primary source of the IUBMB enzyme list. Archived from the original on 10 September 2018. Retrieved 3 November 2018.
External links[edit]
- Enzyme Nomenclature, authoritative website by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology, maintained by G.P. Moss
- Enzyme nomenclature database — by ExPASy
- List of all EC numbers — by BRENDA
- Browse PDB structures by EC number
- Browse SCOP domains by EC number — by dcGO
- Compare EC numbers using EC-Blast Archived 2019-05-30 at the Wayback Machine
The Enzyme Commission number (EC number) is a numerical classification scheme for enzymes, based on the chemical reactions they catalyze.[1] As a system of enzyme nomenclature, every EC number is associated with a recommended name for the corresponding enzyme-catalyzed reaction.
EC numbers do not specify enzymes but enzyme-catalyzed reactions. If different enzymes (for instance from different organisms) catalyze the same reaction, then they receive the same EC number.[2] Furthermore, through convergent evolution, completely different protein folds can catalyze an identical reaction (these are sometimes called non-homologous isofunctional enzymes)[3] and therefore would be assigned the same EC number. By contrast, UniProt identifiers uniquely specify a protein by its amino acid sequence.[4]
Format of number[edit]
Every enzyme code consists of the letters «EC» followed by four numbers separated by periods. Those numbers represent a progressively finer classification of the enzyme. Preliminary EC numbers exist and have an ‘n’ as part of the fourth (serial) digit (e.g. EC 3.5.1.n3).[2]
For example, the tripeptide aminopeptidases have the code «EC 3.4.11.4», whose components indicate the following groups of enzymes:
- EC 3 enzymes are hydrolases enzymes (enzymes that use water to break up some other molecule)
- EC 3.4 are hydrolases that act on peptide bonds
- EC 3.4.11 are those hydrolases that cleave off the amino-terminal amino acid from a polypeptide
- EC 3.4.11.4 are those that cleave off the amino-terminal end from a tripeptide
Top level codes[edit]
Top-level EC numbers[5]| Class | Reaction catalyzed | Typical reaction | Enzyme example(s) with trivial name |
|---|---|---|---|
| EC 1 Oxidoreductases | Oxidation/reduction reactions; transfer of H and O atoms or electrons from one substance to another | AH + B → A + BH (reduced) A + O → AO (oxidized) | Dehydrogenase, oxidase |
| EC 2 Transferases | Transfer of a functional group from one substance to another. The group may be methyl-, acyl-, amino- or phosphate group | AB + C → A + BC | Transaminase, kinase |
| EC 3 Hydrolases | Formation of two products from a substrate by hydrolysis | AB + H2O → AOH + BH | Lipase, amylase, peptidase, phosphatase |
| EC 4 Lyases | Non-hydrolytic addition or removal of groups from substrates. C-C, C-N, C-O or C-S bonds may be cleaved | RCOCOOH → RCOH + CO2 or [X-A+B-Y] → [A=B + X-Y] | Decarboxylase |
| EC 5 Isomerases | Intramolecule rearrangement, i.e. isomerization changes within a single molecule | ABC → BCA | Isomerase, mutase |
| EC 6 Ligases | Join together two molecules by synthesis of new C-O, C-S, C-N or C-C bonds with simultaneous breakdown of ATP | X + Y + ATP → XY + ADP + Pi | Synthetase |
| EC 7 Translocases | Catalyse the movement of ions or molecules across membranes or their separation within membranes | Transporter |
Reaction similarity[edit]
Similarity between enzymatic reactions can be calculated by using bond changes, reaction centres or substructure metrics (formerly EC-BLAST], now the EMBL-EBI Enzyme Portal).[6]
History[edit]
Before the development of the EC number system, enzymes were named in an arbitrary fashion, and names like old yellow enzyme and malic enzyme that give little or no clue as to what reaction was catalyzed were in common use. Most of these names have fallen into disuse, though a few, especially proteolyic enzymes with very low specificity, such as pepsin and papain, are still used, as rational classification on the basis of specificity has been very difficult.
By the 1950s the chaos was becoming intolerable, and after Hoffman-Ostenhof[7] and Dixon and Webb[8] had proposed somewhat similar schemes for classifying enzyme-catalyzed reactions, the International Congress of Biochemistry in Brussels set up the Commission on Enzymes under the chairmanship of Malcolm Dixon in 1955. The first version was published in 1961, and the Enzyme Commission was dissolved at that time, though its name lives on in the term EC Number. The current sixth edition, published by the International Union of Biochemistry and Molecular Biology in 1992 as the last version published as a printed book, contains 3196 different enzymes. Supplements 1-4 were published 1993-1999. Subsequent supplements have been published electronically, at the website of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology.[5] In August 2018, the IUBMB modified the system by adding the top-level EC 7 category containing translocases.[9]
See also[edit]
- List of EC numbers
- List of enzymes
- TC number (classification of membrane transport proteins)
References[edit]
- ^ Webb, E. C. (1992). Enzyme nomenclature 1992: recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the nomenclature and classification of enzymes. Academic Press. ISBN 978-0-12-227164-9.
- ^ a b «ENZYME (Enzyme nomenclature database)». ExPASy. Archived from the original on 21 March 2019. Retrieved 24 April 2019.
- ^ Omelchenko MV, Galperin MY, Wolf YI, Koonin EV (2010). «Non-homologous isofunctional enzymes: a systematic analysis of alternative solutions in enzyme evolution». Biology Direct. 5 (1): 31. doi:10.1186/1745-6150-5-31. PMC 2876114. PMID 20433725.
- ^ Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LS (Jan 2004). «UniProt: the Universal Protein knowledgebase». Nucleic Acids Research. 32 (Database issue): D115–9. doi:10.1093/nar/gkh131. PMC 308865. PMID 14681372.
- ^ a b Moss GP. «Recommendations of the Nomenclature Committee». International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes by the Reactions they Catalyse. Archived from the original on 2018-09-10. Retrieved 2006-03-14.
- ^ Rahman SA, Cuesta SM, Furnham N, Holliday GL, Thornton JM (Feb 2014). «EC-BLAST: a tool to automatically search and compare enzyme reactions». Nature Methods. 11 (2): 171–174. doi:10.1038/nmeth.2803. PMC 4122987. PMID 24412978.
- ^ Hoffman-Ostenhof, O (1953). «Suggestions for a more rational classification and nomenclature of enzymes». Advances in Enzymology and Related Areas of Molecular Biology. Advances in Enzymology and Related Subjects of Biochemistry. Vol. 14. pp. 219–260. doi:10.1002/9780470122594.ch7. ISBN 9780470122594. PMID 13057718.
- ^ Dixon, M; Webb, E.C. (1958). Enzymes. London: Longmans Green. pp. 183–227.
- ^ Tipton, Keith (August 2018). «Enzyme Nomenclature News: Translocases (EC 7): A new EC Class». ExplorEnz: the primary source of the IUBMB enzyme list. Archived from the original on 10 September 2018. Retrieved 3 November 2018.
External links[edit]
- Enzyme Nomenclature, authoritative website by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology, maintained by G.P. Moss
- Enzyme nomenclature database — by ExPASy
- List of all EC numbers — by BRENDA
- Browse PDB structures by EC number
- Browse SCOP domains by EC number — by dcGO
- Compare EC numbers using EC-Blast Archived 2019-05-30 at the Wayback Machine
Классификация ферментов и номенклатура
Первоначально
ферментам давали названия, образуемые
путем добавления окончания — аза к
названию субстрата, на который данный
фермент действует. Так, ферменты,
гидролизующие крахмал (амилон), были
названы амилазами; ферменты, гидролизующие
жиры (липос), -липазами; ферменты,
гидролизующие белки (протеины),
-протеиназами. Позднее ферментам,
катализирующим сходные по типу реакции,
стали давать название, указывающее тип
соответствующей реакции — дегидрогеназы,
оксидазы. декарбоксилазы, ацилазы и
т.д. Многие из этих названий используются
и теперь.
Номенклатура,
введенная Международным биохимическим
союзом (IUB), на первый взгляд кажется
сложной и громоздкой, но зато она является
однозначной. Главный ее принцип состоит
в том, что ферменты называют и классифицируют
в соответствии с типом катализируемой
химической реакции и ее механизмом; это
существенно облегчает систематизацию
данных, относящихся к различным аспектам
метаболизма. Основные черты системы,
введенной IUB, состоят в следующем.
1. Реакции
и ферменты, которые их катализируют,
подразделяются на шесть классов
2. Название фермента
состоит из двух частей: первая часть —
название субстрата (или субстратов);
вторая указывает тип катализируемой
реакции и оканчивается на -аза.
3. Дополнительная
информация, если она необходима для
уточнения, заключается в скобки. Например,
фермент, катализирующий реакцию L-малат
+ NAD+
= Пируват + СО2
+ NADH + Н+
имеет номер 1.1.1.37 и называется L-малат:
NADf
оксидоредуктаза
(декарбоксилирующая).
4. Каждый фермент
имеет кодовый номер по классификации
ферментов (КФ): первая цифра характеризует
класс реакции, вторая — подкласс и третья
— подподкласс. Четвертая цифра указывает
порядковый номер фермента в его
подподклассе. Таким образом, КФ 2.7.1.1
означает, что фермент относится к классу
2 (трансфераза), подклассу 7 (перенос
фосфата) и подподклассу 1 (акцептором
фосфата является спирт). Последняя цифра
обозначает фермент гексокиназу, или
АТР: D-гексозо-б-фос-фотрансферазу, т.е.
фермент, катализирующийперенос фосфата
с АТР на гидроксильную группу атома
углерода в шестом положении глюкозы.
Классификация ферментов
1. Оксидоредуктазы,
ферменты, катализирующие ОВР с участием
двух субстратов
Окисление протекает
как процесс отнятия атомов Н+ от субстрата,
а восстановление – присоединение Н+ к
акцептору. В качестве кофермента
оксидоредуктазы содержат NAD,
NADН,
ФМН, ФАД. Самыми типичным оксидоредуктазами
являются дегидрогеназы. Дегидрогеназы
катализируют
реакции, в которых участвуют группы
СН-ОН, СН-СН, С=О, СН-NH2.
Некоторые подклассы:
Ферменты,
действующие на группу СН-ОН (донор
электронов)
Например:
-
Алкоголь: NAD+
оксидоредуктаза
(алкогольдегидрогеназа)
1.4. Ферменты,
действующие на группу СН-NH2
(донор электронов)
Например:
1.4.1.3. L-глутамат:
NAD(Р)+
оксидоредуктаза (дезаминирующая). Запись
NAD(Р)+
означает, что акцептором электронов
может служить либо NAD+,
либо NAD(Р)+
Самый распространенный
вариант ОВР в клетке состоит в окислении
атомов Н, снятых с субстрата, при
посредстве цитохромной системы.
Цитохромную систему образуют
оксидоредуктазы, имеющие в качестве
простетических групп железопоррфины.
Соединяясь с белками, железопорфины
дают начало семейству хромопротеинов
или цитохромов. Цитохромы образуют
цитохромную систему (b,c,a,a3),
которая передает электроны на кислород,
который соединяется с водородом, образуя
молекулу воды. Из всех цитохромов только
цитохром a,a3
передает электроны на кислород, и поэтому
он называется цитохромоксидазой.
2. Трансферазы,
ферменты катализирующие перенос
функциональной группы (ацильных,
гликозильных, групп, содержащих серу и
фосфор) с одного субстрата на другой
2.3. ацилтрансферазы
Например:
2.3.1.6. Ацетил-СоА:
холин О-ацетилтрансфераза
(холин-ацетилтрансфераза)
2.7. Ферменты,
катализирующие перенос группы, содержащей
фосфор. Например:
2.7.1.1. АТР: D-гексоза
6-фосфотрансфераза (гексозотрансфераза)
3.
Гидролазы,
ферменты, катализирующие гидролиз
эфирных, сложноэфирных, пептидных,
гликозильных связей)
3.1.
Ферменты, действующие на сложноэфирные
связи. Например:
3.1.1.8. Ацилхолин —
ацилгидролаза (псевдохолинэстераза)
Ацилхолин
+ Н20
= холин + кислота
3.2. Ферменты,
действующие на гликозильные соединения.
Например:
3.2.1.23.
β-D-Галактозид-галактогидролаза
(β-галактозидаза)
β-D-Галактозид +
Н20
= спирт + D-галактоза
3.4. Ферменты,
действующие на пептидные связи.
Классификация
(с подразделением на 11 подклассов)
учитывает различия между пептидазами
и протеазами, выделяет ферменты,
гидролизующие дипептиды или более
крупные пептиды, отщепляющие одну или
большее число аминокислот, атакующие
связь на С-или N-конце. Протеиназы в
соответствии с механизмом катализа
подразделяются на сериновые, тиоловые
и металлозависимые. Например:
3.4.21. Сериновые
протеиназы. Например:
химотрипсин,
трипсин, плазмин, факторы свертывания
крови.
3.4.23. Карбоксильные
(кислые) протеиназы. Например,
пепсины А,
В и
С.
4.
Лиазы. Ферменты,
отщепляющие группы от субстратов по
негидролитическому механизму, с
образованием двойных связей.
Это
ферменты, действующие на связи С-С, С-О,
С-N, С-S.
4.1.2.
Альдегид-лиазы.
Например:
4.1.2.7.
Кетозо-1-фосфат-альдолаза (альдолаза)
Кетозо-1-фосфат=дигидроксиацетонфосфат
+альдегид.
4.2. Углерод—кислород
лиазы. Например:
4.2.1.2.
L-малат-гидролиаза (фумараза)
L-малат=Фумарат+Н20
5.
Изомеразы. В
этот класс
включены все ферменты, катализирующие
взаимопревращения оптических,
геометрических и позиционных изомеров.
Некоторые подклассы:
5.2.
цис/транс-изомеразы. Например:
5.2.1.3.
все-транс-Ретиналъ
11-цис-транс-изомераза
(ретинальизомераза)
все-транс-Ретшналь=ll-цис-Ретиналь
5.3.
Ферменты, катализирующие взаимопревращение
альдоз и кетоз. Например:
5.3.1.1.
D-Глицеральдегид-3-фосфаткетол-изомераза
(триозофосфатизомераза)
D-глицеральдегид-З-фосфат=дигидроксиацетонфосфат.
6.
Лигазы,
ферменты, катализирующие соединение
двух молекул, сопряженное с разрывом
пирофосфатной связи АТР. В результате
образуются связи С-О,
С-S, С-N и С-С. Некоторые
подклассы:
6.3. Ферменты,
катализирующие образование связей С-N.
Например:
6.3.1.2.
L—Глутамат:аммиак
лигаза (ADP) (глутаминсинтетаза)
АТР+L-Глутамат+NH+4=ADP+ортофосфат+L-глутамин
6.4.
Ферменты, катализирующие образование
связей С-С.
Например:
6.4.1.2.
Ацетил-СоА:С02
лигаза (ADP)
(ацетил-СоА-карбоксилаза)
АТР+ацетил-СоА+С02=ADP+Рi+малонил-СоА.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Международная классификация ферментов
- Международная классификация ферментов
-
Шифр КФ (Классификация ферментов) или код фермента — это классификационный номер фермента по международной иерархической классификации. Принятая система классифицирует ферменты по группам и индексирует индивидуальные ферменты, что важно для стандартизации исследований.
Классификация ферментов основана и периодически обновляется Комиссией по ферментам (англ. Enzyme commission, отсюда термин «EC number», принятый в англоязычной литературе) при Международном союзе биохимии и молекулярной биологии. Каждый шифр КФ ассоциирован также с рекомендованным названием соответствующего фермента. Классифицировано более 3500 ферментов.
Содержание
- 1 Принцип классификации
- 2 Формат шифра
- 2.1 Класс
- 2.2 Подкласс
- 2.3 Под-подкласс
- 2.4 Код четвертого уровня
- 3 Коды первого уровня
- 4 История
- 5 Ссылки
- 6 См.также
Принцип классификации
Классификация ферментов учитывает реакционную и субстратную специфичности ферментов, а не их белковую структуру. Шифр КФ определяет химическую реакцию, катализируемую ферментом. По этой причине аналогичные ферменты (иногда десятки) из различных организмов имеют один КФ, несмотря на структурные различия.
Иногда различные ферменты одного организма имеют один КФ. Например, панкреатическая липаза и печёночная липаза принадлежат обе к КФ 3.1.1.3 благодаря катализированию одной химической реакции (гидролиз эфирной связи в триглицериде), хотя первый фермент является ферментом пищеварения и работает в кишечнике, а второй относится к ферментам обмена липопротеинов в крови.
Существует база данных UniProt [1] , которая идентифицирует каждый белок по его первичной последовательности. Обе базы данных дополняют друг друга.
Формат шифра
Каждый классификационный номер содержит сокращение КФ и последовательность из четырёх чисел, разделённых точкой, и составляется по определенному принципу. Каждое последующее число представляет собой всё более и более уточняющую классификацию фермента. Так как база данных постоянно обновляется коды могут меняться и некоторые коды могут оставаться незаполненными.
Класс
- Код первого уровня (первое число, может быть от 1 до 6) указывает номер одного из шести главных классов ферментов (см. таблицу Коды первого уровня).
Подкласс
- Код второго уровня (второе число) означает подкласс, характеризующий основные виды субстратов, участвующих в данном типе химических реакций. Например, у трансфераз вторая цифра указывает на природу той группы, которая подвергается переносу, у гидролаз — на тип гидролизуемой связи и т. д.
Под-подкласс
- Код третьего уровня (третье число) определяет более частные подгруппы (под-подклассы), отличающиеся природой химических соединений доноров или акцепторов, участвующих в данной подгруппе реакций. У гидролаз, например, эта цифра уточняет тип гидролизуемой связи, а у лиаз — тип отщепляемой группы и т. д. Первые 3 числа шифра точно определяют тип фермента.
Код четвертого уровня
Наконец, все ферменты, относящиеся к данному под-подклассу, получают свой порядковый номер (четвёртое число в шифре).
- Например, глюкозоксидазе присвоен шифр КФ 1.1.3.4, что означает:
- КФ 1 — Оксидоредуктазы
- КФ 1.1 — Алкогольоксидоредуктазы
- КФ 1.1.3 — Оксидоредуктазы, окисляющие группу CH-OH и восстанавливающие кислород.
- КФ 1.1.3.4 — Оксидоредуктазы, окисляющие глюкозу в присутствие кислорода. Всего известно 3 глюкозоксидазы из разных организмов.
Коды первого уровня
Класс Катализируемая реакция Тип реакции Важнейшие подклассы КФ 1
ОксидоредуктазыОкислительно-восстановительные реакции. Перенос атомов H и O или электронов от одного субстрата на другой AH + B → A + BH (восстановленный)
A + O → AO (окисленный)дегидрогеназа, оксидаза, пероксидаза, редуктаза, монооксидаза, диоксигеназа КФ 2
ТрансферазыПеренос функциональной группы от одного субстрата на другой. Это может быть метильная, ацильная, фосфатная группа или аминогруппа. AB + C → A + BC аминотрансфераза, фосфотрансфераза, C1-трансфераза, гликозилтрансфераза КФ 3
ГидролазыОбразование двух продуктов из одного субстрата в результате гидролиза. AB + H2O → AOH + BH эстераза, гликозил-гидролаза, пептидаза, амидаза КФ 4
Лиазы (синтазы)Негидролитическое добавление или удаление группы к или от субстрата. Образование C-C, C-N, C-O или C-S связи. RCOCOOH → RCOH + CO2 C-O-лиаза, C-S-лиаза, C-N-лиаза, C-C-лиаза КФ 5
ИзомеразыВнутримолекулярная перестановка, то есть изомеризация молекулы субстрата. AB → BA эпимераза, цис-транс-изомераза, внутримолекулярная оксидоредуктаза и др. КФ 6
Лигазы (синтетазы)Соединение двух молекул в результате синтеза новой C-O, C-S, C-N или C-C связи, сопряжённое с одновременным гидролизом АТФ. X + Y+ ATP → XY + ADP + Pi C-O-лигаза, C-S-лигаза, C-N-лигаза, C-C-лигаза История
Схема номенклатуры ферментов была впервые разработана в 1955 году, когда Международный конгресс биохимии в Брюсселе учредил Комиссию по ферментам (Enzyme Commission). Первая версия номенклатуры появилась в 1961 году и включала около 900 ферментов, в версии 1978 года было более 2000 ферментов. Версия 1995 года содержит более 3500 ферментов.
Ссылки
- Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB), Официальный сайт
- Список ферментов
- ↑ ENZYME (Enzyme nomenclature database). ExPASy. Проверено 14 марта 2006.
См.также
- Ферменты
- Список ферментов
- Кластер дифференцировки
Шаблон:Ферменты
Wikimedia Foundation.
2010.
Полезное
Смотреть что такое «Международная классификация ферментов» в других словарях:
-
классификация ферментов — КФ Международная четырехзначная номенклатура ферментов: 1 й знак класс ферментов (1 оксиредуктазы; 2 трансферазы; 3 гидролазы; 4 лиазы; 5 изомеразы; 6 лигазы); 2 й и 3 й знаки подклассы и подподклассы ферментов; 4 й знак порядковый номер в… … Справочник технического переводчика
-
классификация ферментов — Enzyme Classification (EC) классификация ферментов (КФ). Международная четырехзначная номенклатура ферментов: 1 й знак класс ферментов (1 оксиредуктазы; 2 трансферазы; 3 гидролазы; 4 лиазы; 5 изомеразы; 6 лигазы); 2 й и 3 й знаки подклассы и… … Молекулярная биология и генетика. Толковый словарь.
-
Приложение. Список сокращений — a. arteria (ед. число) aa. arteriae (мн. число) ant. anterior b. bursa (ед. число) Bac. Bacillus Bact. Bacterium bb. bursae (мн. число) Ber определитель бактерий Берджи (Bergey’s manual of determinative bacteriology, 8 ed., 1974) BNA Базельская… … Медицинская энциклопедия
-
МКБ-10: Класс III — Международная классификация болезней 10 го пересмотра (МКБ 10) Класс I Некоторые инфекционные и паразитарные болезни Класс II Новообразования Класс III Болезни крови, кроветворных ор … Википедия
-
МКБ-10: Класс III. Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм — Международная классификация болезней 10 го пересмотра (МКБ 10) Класс I Некоторые инфекционные и паразитарные болезни Класс II Новообразования Класс III Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм Класс … Википедия
-
МКБ-10: Класс XX — Международная классификация болезней 10 го пересмотра (МКБ 10) Класс I Некоторые инфекционные и паразитарные болезни Класс II Новообразования Класс III Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный ме … Википедия
-
МКБ-10: Код Y — Международная классификация болезней 10 го пересмотра (МКБ 10) Класс I Некоторые инфекционные и паразитарные болезни Класс II Новообразования Класс III Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм Класс … Википедия
-
Хронический панкреатит — МКБ 10 K86.086.0 K86.186.1 МКБ 9 577.1 … Википедия
-
Инфекцио́нные боле́зни — (позднелат. infectio заражение) группа болезней, которые вызываются специфическими возбудителями, характеризуются заразительностью, циклическим течением и формированием постинфекционного иммунитета. Термин «инфекционные болезни» был введен… … Медицинская энциклопедия
-
МКБ-10: Класс IV — Список классов Международной классификации болезней 10 го пересмотра Класс I. Некоторые инфекционные и паразитарные болезни Класс II. Новообразования Класс III. Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный… … Википедия
Ферменты
Катализаторы — это вещества, ускоряющие химические реакции; в ходе реакции они претерпевают физические изменения, но по ее завершении возвращаются в исходное состояние. Ферменты — это биологические катализаторы. Большая часть ферментов является белками. Существуют рибозимы — ферменты, являющиеся молекулами РНК. К рибозимам относится РНК большой субъединицы рибосомы, осуществляющий биосинтез белка, РНКаза Р (англ.), осуществляющая разрезание предшественников тРНК, и интроны авотосплайсинга в митохондриях некоторых инфузорий.
В отличие от небелковых катализаторов (Н+, ОН—, ионы металлов) каждый фермент способен катализировать лишь очень небольшое число реакций, часто только одну. Таким образом, ферменты представляют собой реакционно-специфические катализаторы. Практически все биохимические реакции катализируются ферментами.
Классификация ферментов и их номенклатура
Первоначально ферментам давали названия, образуемые путем добавления окончания -аза к названию субстрата, на который данный фермент действует. Так, ферменты, гидролизующие крахмал (амилон), были названы амилазами; ферменты, гидролизующие жиры (липос), — липазами; ферменты, гидролизуюшие белки (протеины), протеиназами. Позднее ферментам, катализирующим сходные по типу реакции, стали давать название, указывающее тип соответствующей реакции — дегидрогеназы, оксидазы, декарбоксилазы, ацилазы и т. д. Многие из этих названий используются и теперь. Номенклатура, введенная Международным биохимическим союзом (IUB), на первый взгляд кажется сложной и громоздкой, но зато она является однозначной. Главный ее принцип состоит в том, что ферменты называют и классифицируют в соответствии с типом катализируемой химической реакции и ее механизмом; это существенно облегчает систематизацию данных, относящихся к различным аспектам метаболизма. Основные черты системы, введенной IUB, состоят в следующем.
1. Реакции и ферменты, которые их катализируют, подразделяются на шесть классов, в каждом из которых имеется несколько подклассов (от четырех до 13).
2. Название фермента состоит из двух частей: первая часть — название субстрата (или субстратов); вторая указывает тип катализируемой реакции и оканчивается на -аза.
3. Дополнительная информация, если она необходима для уточнения, заключается в скобки. Например, фермент, катализирующий реакцию L-малат + NAD+ = Пируват + СО2 + NADH + Н +, имеет номер 1.1.1.37 и называется L-малат: NAD+ оксидоредуктаза (декарбоксилирующая).
4. Каждый фермент имеет кодовый номер по классификации ферментов (КФ): первая цифра характеризует класс реакции, вторая — подкласс и третья — подподкласс. Четвертая цифра указывает порядковый номер фермента в его подподклассе. Таким образом, КФ 2.7.1.1 означает, что фермент относится к классу 2 (трансфераза), подклассу 7 (перенос фосфата) и подподклассу 1 (акцептором фосфата является спирт). Последняя цифра обозначает фермент гексокиназу, или АТФ: D-гексозо-6-фосфотрансферазу т. е. фермент, катализирующий перенос фосфата с АТФ на гидроксильную группу атома углерода в шестом положении глюкозы. Ниже представлены все шесть классов ферментов и некоторые конкретные примеры. В скобках указано рекомендуемое название.
Ферменты делятся на шесть классов в соответствии с типом катализируемой реакции:
1. Оксидоредуктазы. Ферменты, катализирующие окислительно-восстановительные реакции с участием двух субстратов, S и Ś:
Sвосст + Śокисл =Sокисл+Śвосст
Катализируют реакции, в которых участвуют такие группы, как СН — ОН, СН — СН, С = О, СН — NH2 и — СН — NH — . Некоторые подклассы:
1.1. Ферменты, действующие на группу СН — ОН (донор электронов). Например:
1.1.1.1. Алкоголь: NAD+оксидоредуктаза [алкогольдегидрогеназа]
Спирт + NAD+ = Альдегид или кетон + NADH + Н+.
1.4. Ферменты, действующие на группу СН — NH2 (донор электронов). Например:
1.4.1.3. L-Глутамат: NAD (P) + оксидоредуктаза (дезаминирующая) [глутаматдегидрогеназа из печени животных]. Запись NAD (P) + означает, что акцептором электронов может служить либо NAD+, либо NADP+.
L-Глутамат + Н2О + NAD (P) + = = α-Кетоглутарат + NH+4 + NAD (P) H + Н+.
2. Трансферазы. Ферменты, катализирующие перенос группы G (отличной от атома водорода) с субстрата S на субстрат Ś:
S — G + Ś = Ś — G + S.
Катализируют перенос одноуглеродных групп, альдегидных или кетонных остатков, а также ацильных, алкильных, гликозильных групп и групп, содержащих фосфор и серу. Некоторые подклассы:
2.3. Ацилтрансферазы. Например:
2.3.1.6. Ацетил-СоА: холин О-ацетилтрансфераза [холин-ацетилтрансфераза]
Ацетил-СоА + Холин = СоА + О-Ацетилхолин.
2.7. Ферменты, катализирующие перенос группы, содержащей фосфор. Например:
2.7.1.1. АТР: D-гексоза 6-фосфотрансфераза [гексокиназа]
АТР + D-Гексоза = ADP + D-Гексозо-6-фосфат.
3. Гидролазы. Ферменты, катализирующие гидролиз эфирных, сложноэфирных, пептидных и гликозидных связей, кислотных ангидридов, связей С — С, С-галоида и Р — N.
S-Ś + H2O = S + Ś
Например:
3.1. Ферменты, действующие на сложноэфирные связи. Например: 3.1.1.8. Ацилхолин — ацилгидролаза [псевдохолинэстераза]
Ацилхолин + Н2О = Холин + Кислота.
3.2. Ферменты, действующие на гликозидные соединения. Например:
3.2.1.23. β-D-Галактозид — галактогидролаза [β-галактозидаза]
β-D-Галактозид + Н2О = Спирт + D-Галактоза.
3.4. Ферменты, действующие на пептидные связи.
Классификация (с подразделением на 11 подклассов) учитывает различия между пептидазами и протеазами, выделяет ферменты, гидролизующие дипептиды или более крупные пептиды, отщепляющие одну или большее число аминокислот, атакующие связь на С- или N-конце. Протеиназы в соответствии с механизмом катализа подразделяются на сериновые, тиоловые и металлозависимые. Например:
3.4.21. Сериновые протеиназы. Например: Химотрипсин, трипсин, плазмин, факторы свертывания крови IХа и ХIа.
3.4.23. Карбоксильные (кислые) протеиназы. Например, пепсины А, В и С.
4. Лиазы. Ферменты, отщепляющие группы от субстратов по негидролитическому механизму, с образованием двойных связей.
Рисунок 79. Схема работы лиаз
Ферменты, действующие на связи С — С, С — О, С — N, С — S и С — галоид. Некоторые подгруппы:
4.1.2. Альдегид-лиазы. Например:
4.1.2.7. Кетозо-1 -фосфат-альдолаза [альдолаза]
Кетозо-1-фосфат = Дигидроксиацетонфосфат +Альдегид.
4.2. Углерод — кислород лиазы. Например:
4.2.1.2. L-малат — гидролиаза [фумараза]
L-малат = Фумарат + Н2О.
5. Изомеразы. В этот класс включены все ферменты, катализирующие взаимопревращения оптических, геометрических и позиционных изомеров.
S D↔ S L
Некоторые подклассы:
5.2. Цистранс-изомеразы. Например:
5.2.1.3 11 -цис-транс-изомераза [ретинальизомераза]
5.3. Ферменты, катализирующие взаимопревращение альдоз и кетоз. Например:
5.3.1.1. D-Глицеральдегид-3-фосфаткетол-изомераза [триозофосфатизомераза]
D-Глицеральдегид-3-фосфат = Дигидроксиацетонфосфат.
6. Лигазы. (от лат. лигаре — связывать). Ферменты, катализирующие соединение двух молекул, сопряженное с разрывом пирофосфатной связи АТР или подобного соединения. В этот класс включены ферменты, катализирующие реакции, в ходе которых образуются связи С — О, С — S, С — N и С — С.
S + Ś = S-Ś
Некоторые подклассы:
6.3. Ферменты, катализирующие образование связей С — N. Например:
6.3.1.2. L-Глутамат: аммиак лигаза (ADP) [глутаминсинтетаза]
АТР + L-Глутамат + NH4+ = ADP + Ортофосфат + L-Глутамин.
6.4. Ферменты, катализирующие образование связей С — С. Например:
6.4.1.2. Ацетил-СоА: СО2 лигаза (ADP) [ацетил-Со А — карбоксилаза]
АТР + Ацетил-СоА + СО2 = ADP + Р; + Малонил-СоА.
Коферменты
Холофермент — полностью функциональная молекула фермента
Апофермент — белковая часть фермента
Кофермент — специфическое термостабильное низкомолекулярное органическое соединение, которое участвует в ферментативной реакции в качестве дополнительного субстрата, принимающего на себя группировки в одного субстрата, и передавая их на другой. По сути являющееся постоянно связанным с каталитическим центром вторым субстратом.
Если фермент простой белок, то холофермент = апофермент.
Если фермент сложный белок, холофермент = апофермент + кофермент.
В ходе реакции кофермент претерпевает химические изменения, в точности противоположные изменениям, которые происходят в субстрате. Например, в окислительно-восстановительных дегидрогеназных реакциях молекула субстрата окисляется, а молекула кофермента восстанавливается. Подобным же образом в реакциях переаминирования пиридоксальфосфат выступает и как второй субстрат в двух связанных реакциях, и как переносчик аминогруппы между различными α-амино- и α-кетокислотами.
Вторая причина, по которой кофермент можно считать равноправным участником реакции, заключается в том, что именно его участие может иметь фундаментальное физиологическое значение. Например, работа мышцы в анаэробных условиях сопровождается превращением пирувата в лактат. Но в этом случае важны совсем не лактат и не пируват: предназначением реакции является превращение NADH в NAD+. В отсутствие NAD+ гликолиз продолжаться не может и анаэробный синтез АТР (а, следовательно, и работа мышцы) прекращается. Восстановление пирувата до лактата в анаэробных условиях обеспечивает окисление NADH в NAD+, необходимый для синтеза АТР. Функцию образования NAD+ могут выполнять и другие реакции.
Значение этого процесса становится очевидным, если перейти от животных к другим формам жизни. У бактерий и дрожжей, растущих в анаэробных условиях, вещества, образующиеся из пирувата, служат окислителями для NADH, при этом сами они восстанавливаются.
Кофермент может быть связан с апоферментом ковалентными или нековалентными связями. К числу реакций, требующих присутствия коферментов, относятся окислительно-восстановительные реакции, реакции переноса групп и изомеризации, а также реакции конденсации (по системе IUB это классы 1, 2, 5 и 6). Реакции расщепления, например гидролитические реакции, катализируемые пищеварительными ферментами, протекают в отсутствие кофермента.
Можно предложить следующую классификацию коферментов (Таблица 3):
Таблица 3. Классификация коферментов
Механизмы действия ферментов
Все химические реакции происходят в соответствии с теорией столкновений.
Кинетическая теория или теория столкновений
Кинетическая теория, или теория столкновений, основывается на двух ключевых положениях.
1. Чтобы столкновение было продуктивным (т. е. приводило к протеканию реакции), реагирующие молекулы должны обладать энергией, достаточной для преодоления энергетического барьера.
Отсюда следует, что при наличии у реагирующих молекул достаточной энергии все факторы, повышающие частоту их столкновений, будут повышать скорость реакции. И наоборот, факторы, понижающие частоту столкновений молекул или их кинетическую энергию, снижают скорость реакции.
Если не все молекулы в популяции обладают энергией, достаточной для осуществления реакции, то повышение температуры, сопровождающееся увеличением кинетической энергии молекул, приведет к повышению скорости реакции. В случае А ни одна из молекул, в случае Б — часть, а в случае В — все молекулы обладают кинетической энергией, достаточной для преодоления энергетического барьера.
Эта энергия столкновений получила название энергии перехода. То есть молекулы обладающие дополнительной энергией, сталкиваясь, вступают в реакцию.
Как и любой катализатор, ферменты понижают энергию перехода. В обоих случаях величина свободной энергии образования переходного состояния характеризует энергетический барьер полной реакции (Рисунок 80). Но энергетический барьер реакции, проходящей через переходное состояние [Y-R-•Х], ниже, чем энергетический барьер реакции, проходящей через переходное состояние [Y-R-X]. В приведенном выше примере [Y-R-X] представляет собой переходное состояние некатализируемой реакции, тогда как [Y-R-X] b — это переходное состояние катализируемой реакции. Все катализаторы, включая и ферменты, уменьшают свободную энергию образования переходного состояния ΔG0. Заметим далее, что на величину ΔG0 катализатор не влияет: изменение свободной энергии полной реакции не зависит от присутствия катализаторов. Константа равновесия химической реакции является функцией изменения стандартной свободной энергии этой реакции:
ΔG0 = -RT ln Keq
Отсюда следует, что ферменты и другие катализаторы не влияют на константу равновесия реакции.
2. Для протекания реакции молекулы должны сталкиваться друг с другом, т. е. сближаться на расстояния, достаточные для образования связей. Для этого можно повысить концентрацию реагирующих веществ, а можно объединить их в одной точке пространства, увеличив концентрацию локально, что часто происходит в ходе функционирования катализаторов.
Такое ее представление подчеркивает три важных признака ферментативных реакций переноса групп.
1. Каждая полуреакция сопровождается и разрывом, и образованием ковалентной связи.
2. Фермент является равноправным реагентом, таким же, как D — G и А.
3. В то время как в полной реакции фермент выполняет функцию катализатора (т. е. он требуется лишь в следовых количествах и возвращается в исходное состояние по окончании реакции), в каждой из полуреакций фермент выступает как стехиометрический реагент (т. е. реагирует с другими реагентами в молярном отношении 1:1). Многие другие биохимические реакции можно рассматривать как частные случаи реакций переноса, в которых отсутствуют либо А, либо D, либо оба реагента. Так, реакцию изомеризации (например, взаимопревращение глюкозо-6-фосфата и глюкозо-1-фосфата) можно представить как реакцию переноса, в которой отсутствуют D и А.
Рисунок 80. Механизм действия ферментов. А — график изменения энергии в ходе химической реакции, Б — локальное изменение концентрации в ходе работы ферментов
Из всего сказанного выше ясно, что для протекания реакции необходимо, чтобы все участвующие в ней реактанты сближались на расстояния, достаточные для образования (или для разрыва) связей, т. е. сталкивались друг с другом. В химии гомогенных растворов концентрация реагирующих молекул в отсутствие катализаторов считается постоянной во всем растворе. Однако в присутствии катализатора это условие перестает соблюдаться. Для эффективной работы катализатор должен иметь на своей поверхности участки связывания реагирующих молекул. Такое связывание представляет собой обратимый процесс, однако равновесие сильно сдвинуто в сторону образования комплекса. Качественно это можно представить следующим образом:
Реактант + Катализатор = Комплекс реактанта с катализатором.
Прочность комплекса реактанта R и катализатора С можно охарактеризовать количественно с помощью константы диссоциации комплекса R — С (Кd) Отсюда можно получить одно важное следствие: связывание реактанта с катализатором приводит к заметному повышению локальной концентрации реагента по сравнению с его концентрацией во всем растворе.
Если катализатор биомолекулярной реакции (идущей с участием двух реактантов) связывает оба реактанта, то локальная концентрация каждого из них повышается, причем степень этого повышения зависит от сродства катализатора к данному реактанту (Kd). Одним из ключевых факторов, позволяющих ферменту служить катализатором, является его способность эффективно связывать один или (чаще) оба реактанта, участвующие в бимолекулярной реакции, что приводит к повышению локальной концентрации реактантов и, следовательно, к локальному повышению скорости реакции. То обстоятельство, что ферменты по сравнению с большинством небелковых катализаторов необычайно эффективны и высокоизбирательны, требует дальнейшего объяснения. Чтобы понять эти отличительные свойства ферментов, мы должны ввести понятие активного, или каталитического, центра.
Каталитический центр
Размеры белков намного превышают размеры низкомолекулярных субстратов, в связи с чем возникло представление о том, что в катализе участвует лишь ограниченная область молекулы фермента. Эту область мы и называем каталитическим центром. Каталитический центр — это несколько аминокислотных остатков, формирующих особую часть белка, в которой происходит ферментативная реакция. Вначале было непонятно, почему молекулы ферментов столь велики, если только часть их структуры участвует в связывании субстрата и непосредственно в катализе. Однако, как показал анализ трехмерной структуры ферментов, с субстратом взаимодействует намного большая часть белковой молекулы, чем предполагалось ранее. Если еще учесть включение в работу ферментов аллостерических центров такого же размера не приходится удивляться объемности ферментов.
Связь фермента с субстратом
Большинство субстратов образует, по меньшей мере, три связи с ферментом. Благодаря такой «трехточечной фиксации» симметричная молекула может проявлять асимметрию. Чтобы это пояснить, представим область фермента, связывающую субстрат, как участок плоской поверхности (хотя, как мы вскоре увидим, субстратсвязывающая «площадка» фермента редко бывает плоской, а возможно, и не бывает совсем). Если молекула субстрата может подойти к этому участку только с одной стороны, и взаимодействовать могут только комплементарные структуры субстрата и фермента (в реальных ферментах оба этих условия соблюдаются), то молекула субстрата может связываться с ферментом единственным способом, даже если группы 1 и 3 идентичны. Перебирая мысленно все возможные пространственные ориентации молекулы субстрата, можно убедиться, что с тремя точками плоской поверхности (с одной и той же стороны) молекула может связаться только в одной ориентации. Отсюда следует, что группы 1 и 3, хотя они и идентичны, при связывании с ферментом становятся неэквивалентными из-за различия в их окружении. Химические изменения будут происходить только с группой 1, но не с группой 3 (или наоборот). Обобщая эти рассуждения, можно объяснить теперь, почему ферментативное восстановление оптически неактивного пирувата приводит к образованию именно L-, а не D, L-лактата. Именно такой способ присоединения обеспечивает специфичность фермента.
Рисунок 81. Механизм связывания минимум в 3-х точках
Специфичность ферментов
Специфичность ферментов — это способность фермента катализировать одну и только одну специфическую реакцию является, пожалуй, наиболее важным его свойством. Благодаря этому скорости специфических метаболических процессов могут регулироваться путем изменения каталитической активности специфических ферментов. Правда, многие ферменты катализируют реакции одного типа (перенос фосфата, окислительно-восстановительные реакции и т. д.), субстратами при этом является небольшое число структурно сходных соединений. Реакции с альтернативными субстратами происходят в тех случаях, когда эти субстраты присутствуют в высоких концентрациях. Протекают ли в живых организмах все реакции, возможные при участии данного фермента, зависит от относительной концентрации альтернативных субстратов в клетке и относительного сродства фермента к этим субстратам.
Оптическая специфичность ферментов
За исключением эпимераз (рацемаз), которые катализируют взаимопревращение оптических изомеров, ферменты в общем случае проявляют абсолютную оптическую специфичность, по крайней мере, по отношению к одному из участков молекулы субстрата. Так, ферменты гликолитического и прямого окислительного пути катализируют превращения только D-, но не L-фосфосахаров. За единичными исключениями (например, почечная оксидаза D-аминокислот) большинство ферментов млекопитающих катализирует превращение только L-изомеров аминокислот.
Оптическая специфичность может относиться как к фрагменту молекулы, так и к молекуле в целом. Иллюстрацией служит специфичность гликозидаз. Эти ферменты катализируют гидролиз гликозидных связей между сахаром и спиртовой группой: они высоко-специфичны как к сахарному фрагменту, так и к характеру гликозидной связи (α или β), но относительно неспецифичны к спиртовому фрагменту молекулы.
Группоспецифичность ферментов
Литические ферменты действуют на специфические химические группировки: гликозидазы — на гликозидные связи, пепсин и трипсин — на пептидные связи. Действие этих ферментов распространяется на большое число субстратов, что позволяет организму обойтись небольшим числом пищеварительных ферментов — иначе их потребовалось бы намного больше.
Отдельные литические ферменты отличаются более высокой группоспецифичностью. Так, химотрипсин гидролизует преимущественно пептидные связи, в которых карбоксильная группа принадлежит ароматическим аминокислотам — фенилаланину, тирозину или триптофану. Карбоксипептидазы и аминопептидазы отщепляют аминокислоты по одной с карбоксильного или с амино-конца соответственно.
Некоторые оксидоредуктазы способны использовать в качестве акцепторов электронов и NAD+, и NADPH, но большая их часть использует только один из них. Обобщая, можно сказать, что оксидоредуктазы млекопитающих, которые участвуют в биосинтетических процессах (например, в синтезе жирных кислот или стероидов), обычно используют в качестве восстановителя NADPH, тогда как в катаболизме участвуют оксидоредуктазы использующие NADH.
Модель «ключ — замок»
Первоначальная модель каталитического центра, предложенная Эмилем Фишером, трактовала взаимодействие субстрата и фермента по аналогии с системой «ключ — замок». Эта модель, которую иногда называют моделью «жесткой матрицы», не утратила своего значения для понимания некоторых свойств ферментов, например их способности к строго определенному связыванию двух или большего числа субстратов, или для объяснения кинетики насыщения субстратом (Рисунок 82 А).
Рисунок 82. Модели взаимодействия фермента и субстрата. А-модель «ключ-замок», Б-модель индуцированного соответствия
Модель индуцированного соответствия
Недостатком модели Фишера является подразумеваемая в ней жесткость каталитического центра. Более общий характер имеет модель индуцированного соответствия (Рисунок 82 Б), предложенная Кошландом. Эта модель основывается на весьма убедительных экспериментальных данных. Ее существенной чертой является гибкость каталитического центра. В модели Фишера каталитический центр считается заранее подогнанным под форму молекулы субстрата. В модели же индуцированного соответствия субстрат индуцирует конформационные изменения фермента, и лишь в результате этих изменений аминокислотные остатки и другие группы фермента принимают пространственную ориентацию, необходимую для связывания субстрата и катализа. При этом другие аминокислотные остатки могут погрузиться в глубь молекулы фермента. Субстрат по мере сближения с ферментом индуцирует в последнем конформационные изменения, в результате которых соответствующие группы занимают положение, необходимое для связывания субстрата и для катализа. Одновременно меняется пространственное расположение других остатков — Lys и Met теперь оказываются сближенными. Аналоги субстрата тоже могут вызывать конформационные изменения, но не все из них являются «правильными». При связывании истинного субстрата (А) все группы занимают нужное положение. При связывании же аналога субстрата — более объемного или, наоборот, меньшего по размеру — индуцируется неправильное расположение этих групп.
Факторы, влияющие на каталитическую активность
В лабораторных условиях на скорость ферментативных реакций влияют следующие факторы:
концентрация фермента
концентрация субстрата,
температура,
рН,
присутствие ингибиторов.
Температура
В некотором ограниченном интервале температур скорость ферментативной реакции повышается с ростом температуры. Коэффициент, указывающий, во сколько раз повышается скорость реакции при повышении температуры на 10°, называется температурным коэффициентом и обозначается Q10. Для многих биологических реакций при повышении температуры на 10° скорость удваивается (Q10 = 2) и, аналогично, при понижении температуры на 100 уменьшается вдвое. Многие физиологические процессы (например, скорость сокращения изолированной сердечной мышцы) тоже характеризуются коэффициентом Q10, близким к двум. Однако при некой оптимальной температуре скорость реакции максимальна. Повышение скорости реакции по мере приближения к оптимальной температуре объясняется увеличением кинетической энергии реагирующих молекул. При дальнейшем повышении температуры кинетическая энергия молекулы фермента становится достаточной для разрыва связей, поддерживающих вторичную структуру фермента в нативном, каталитически активном состоянии (происходит тепловая денатурация фермента). Вторичная и третичная структура фермента разрушается, что сопровождается потерей каталитической активности. Денатурация фермента приводит к уменьшению скорости реакции из-за уменьшения концентрации фермента (катализатора).
Для большинства ферментов оптимальная температура равна или выше той температуры, при которой в норме находятся клетки. Для ферментов микроорганизмов, адаптировавшихся к обитанию в природных горячих источниках, оптимальная температура может быть близка к точке кипения воды.
рН
Умеренные изменения рН оказывают влияние на ионное состояние фермента, а зачастую и субстрата. Как показывают измерения ферментативной активности при различных рН, оптимум активности находится обычно между рН 5,0 и 9,0. Вместе с тем отдельные ферменты, например пепсин, активны при значениях рН, лежащих далеко за пределами этого интервала.
Зависимость активности от рН определяется следующими факторами.
1. Денатурацией фермента при очень высоких или очень низких рН. При изменении рН ферменты могут претерпевать конформационные изменения. Для поддержания активной третичной или четвертичной структуры может оказаться необходимым присутствие определенного заряда на группе, удаленной от области связывания субстрата; именно такая ситуация наблюдается в случае гемоглобина. Если заряд этой группы изменится, могут произойти частичное развертывание белковой цепи, или, наоборот, компактизация молекулы, или же ее диссоциация на протомеры — во всех случаях с потерей активности.
2. Изменением величины заряда молекул субстрата или фермента. Активность фермента может изменяться в результате изменений либо его структуры, либо заряда функциональных остатков, участвующих в катализе или связывании субстрата. Рассмотрим для примера взаимодействие отрицательно заряженного фермента (Enz—) с положительно заряженным субстратом (SH+). При низких рН происходит протонирование Enz—, при высоких рН — депротонирование субстрата. Поскольку, взаимодействовать друг с другом могут только SH+ и Enz—, при крайних значениях рН эффективная концентрация Enz— или SH+ будет низкой, что приведет к снижению скорости реакции.
Концентрация фермента
Во многих случаях бывает недостаточно знать, что данный фермент присутствует в системе, нужна еще информация о его количестве. При определенных условиях скорость ферментативной реакции прямо пропорциональна количеству фермента. Это бывает не всегда, что можно проиллюстрировать на примере прямой реакции, идущей в равновесных условиях. Даже если мы знаем, что прямая реакция действительно идет, нам будет казаться, что скорость ее равна нулю, поскольку с такой же скоростью идет и обратная реакция. Когда же ферментативная реакция только начинается, продукт еще практически отсутствует и обратная реакция не идет. Кроме того, на начальной стадии реакции концентрация субстрата соответствует его исходному количеству. Поэтому скорость в начале реакции, т. е. ее начальная скорость (г), будет прямо пропорциональна концентрации фермента [Enz].
Фермент является реактантом, который соединяется с субстратом, образуя фермент-субстратный комплекс Enz — S, распадающийся на свободный фермент и продукт Р. В простейшей форме можно записать
Таким образом, концентрация фермента не оказывает влияния на константу равновесия. Кeq не зависит от того, каким образом достигается равновесие — с участием фермента или без него (вспомним о величине ΔG°). Фермент меняет путь, по которому идет реакция, но не конечные (равновесные) концентрации реагентов и продуктов, от которых зависят Кeq и ΔG°.
Таким образом концентрация фермента оказывает влияние на скорость ферментативной реакции только на начальных этапах ферментативной реакции, следовательно данный механизм воздействия на скорость ферментативной реакции не эффективен, поэтому не используется. С другой стороны можно использовать малые количества ферментов для полного прохождения реакции.
Концентрация субстрата
При изучении зависимости скорости ферментативной реакции от концентрации субстрата были рассмотрены ферментативные реакции, в которых имеется единственный субстрат и единственный продукт. Такая ситуация действительно наблюдается для некоторых ферментативных реакций, однако большинство из них протекает с участием двух и более субстратов и продуктов. Это, однако, нисколько не снижает ценности дальнейших рассуждений. То, что справедливо для одного субстрата, остается верным и для двух.
При увеличении концентрации субстрата [S] и сохранении всех остальных условий начальная скорость v (скорость, измеряемая в период, когда израсходована очень малая доля субстрата) будет повышаться до максимальной величины Кmах, после чего останется постоянной.
По мере повышения концентрации субстрата скорость будет расти до тех пор, пока не произойдет насыщения фермента субстратом. Измеренная в таких условиях начальная скорость уже не будет возрастать при дальнейшем повышении концентрации субстрата. Отметим, что субстрат обычно берут в значительном молярном избытке по отношению к ферменту. Например, если фермент с мол. массой 100000 взаимодействует с субстратом с мол. массой 100 и оба присутствуют в концентрации 1 мг/мл, то на каждый моль фермента будет приходиться 1000 молей субстрата. Более реальными являются следующие величины:
[Enz] = 0,1 мкг/мл = 10—9 М, [S] = 0,1 мг/мл = 10—3 М,
т. е. молярный избыток субстрата по отношению к ферменту составляет 106.
Даже если [S] уменьшить в 100 раз, его концентрация все еще в 10000 раз будет превышать концентрацию фермента. В точках А и В в комплексе с субстратом находится лишь часть молекул фермента, хотя молекул субстрата намного больше, чем молекул фермента. Это происходит потому, что константа равновесия реакции Enz + S +± Enz — S (образование комплекса Enz — S) хотя и велика, но конечна. Таким образом, в точках А и В повышение или понижение [S] будет приводить к увеличению или уменьшению доли молекул Enz, связанных с S (т. е. доли молекул Enz — S), и v будет зависеть от [S]. В точке С практически все молекулы фермента связаны с субстратом, и дальнейшее повышение [S], хотя и повысит частоту столкновений Enz с S, не сможет привести к повышению скорости реакции — среди молекул фермента уже не будет таких, которые были бы свободны для реакции с субстратом.
Случай В представляет особый теоретический интерес, поскольку при этом ровно половина молекул фермента насыщена субстратом. Соответственно скорость равна половине максимальной скорости (VmdJ2), достижимой при данной концентрации фермента.
Уравнение Михаэлиса-Ментен
Уравнение Михаэлиса — Ментен описывает зависимость скорости ферментативной реакции от концентрации субстрата. Фермент Е соединяется с субстратом S, образуя ES-комплекс; константа скорости этого процесса — k1. Судьба ES-комплекса складывается двояко: он может либо диссоциировать на Е и S с константой скорости k2, либо подвергаться дальнейшему превращению, образуя продукт Р, с константой скорости k3(Рисунок 83—1). Постулируется, что продукт реакции Р не превращается в исходный субстрат S; это условие соблюдается на начальной стадии реакции, пока концентрация продукта не достигает ощутимого количества.
Как связана скорость катализа с концентрацией субстрата и фермента и скоростями отдельных этапов реакции? Начнем с того, что скорость ферментативной реакции равна произведению концентрации комплекса ES на константу k3 (Рисунок 83—2).
Выразим [ES] через известные величины. Скорости образования и распада ES (Рисунок 83—3,4).
Определим скорость катализа в стационарных условиях. Стационарные условия характеризуются тем, что концентрация промежуточных продуктов остается постоянной, тогда как концентрации исходных и конечных продуктов меняются. Это имеет место в том случае, когда скорость синтеза ES-комплекса оказывается равной скорости его распада. Если левые части равенств равны между собой, то равны и правые (Рисунок 83—5).
Преобразуем это уравнение (Рисунок 83—6).
Уравнение можно упростить, если ввести новую константу Км, называемую константой Михаэлиса (Рисунок 83—7).
Введем Км в уравнение (Рисунок 83—8).
Рассмотрим числитель в последнем выражении. Концентрация несвязанного субстрата [S] практически равна общей концентрации субстрата при условии, что концентрация фермента значительно ниже концентрации субстрата. Концентрация несвязанного фермента (Е) равна общей концентрации фермента Ет минус концентрация комплекса ES (Рисунок 83—9).
Подставим это выражение в уравнение (Рисунок 83—10).
Решение уравнения относительно [ES] дает (Рисунок 83—11) или (Рисунок 83—12).
Подставим это выражение в уравнение скорости реакции (Рисунок 83—13).
После преобразования получаем окончательный вариант уравнения Михаэлиса — Ментен (Рисунок 83—15).
Рисунок 83. Вывод уравнения Михаэлиса-Ментен
Графическое определение константы Михаэлиса Км
Концентрация субстрата, при которой скорость составляет половину максимальной, обозначается через Км и называется константой Михаэлиса. Ее можно определить из графика зависимости v от [S]. Обратите внимание, что Км имеет размерность молярной концентрации.
Рисунок 84. График зависимости скорости ферментативной реакции от концентрации субстрата (описывается уравнением Михаэлиса-Ментен)
Когда [S] приближается к Км, v становится весьма чувствительной к изменению [S]; в этой области фермент работает со скоростью, равной половине максимальной. Многие ферменты характеризуются такими значениями Км, которые примерно соответствуют физиологическим концентрациям их субстратов.
Уравнение Михаэлиса — Ментен описывает поведение многих ферментов при изменении концентрации субстратов (Рисунок 84). Используя это уравнение, зависимость начальной скорости ферментативной реакции от [S] и Км можно проиллюстрировать на следующих конкретных примерах.
1. [S] много меньше Км (точка А).
В этом случае величину [S] в знаменателе можно опустить, и он будет практически равен Км. Отношение двух констант, Vmax и Км, можно заменить новой константой К. Таким образом, имеем:
Другими словами, когда концентрация субстрата значительно ниже той, при которой скорость реакции составляет половину максимальной (т. е. значительно меньше Км), начальная скорость v пропорциональна концентрации субстрата [S].
2. [S] много больше Км (точка С). В этом случае член Км в знаменателе можно опустить, т. е. Это означает, что при концентрации субстрата [S], намного превышающей Kм начальная скорость v равна максимальной Vmax
3. [S] = Км (точка В). Это означает, что при концентрации субстрата, равной Км, начальная скорость реакции v составляет половину максимальной. Отсюда же следует способ оценки Км: надо экспериментально определить концентрацию субстрата, при которой начальная скорость равна половине максимальной.
Как видно из рисунка график, описываемый уравнением Михаэлиса-Ментен, является кривой, чтобы кривая была точной необходимо максимальное количество точек для ее построения, следовательно, необходимо множество замеров скорости реакции с разными концентрациями субстрата. Тогда как для построения прямой требуется всего 3—5 точек, следовательно 3—5 замеров, именно поэтому было проведено преобразование уравнения Михаэлиса-Ментен (Рисунок 85, 86). График делается в обратных координатах 1/V и 1/ [S]. Для преобразования необходимо 1 разделить на уравнение:
Рисунок 85. Преобразование Лайнуивера-Бэрка
Как видно из преобразования, данное уравнение можно свести к уравнению прямой у = ах + b, где y = l/V; x = 1/ [S]. Данное преобразование по фамилиям авторов получило название — преобразование Лайнуивера-Бэрка.
Используя график Лайнуивера-Бэрка на практике для оценки Км, иногда сталкиваются с тем, что почти все точки оказываются в области низких концентраций субстрата. Это происходит в том случае, когда измерения проводят через равные интервалы [S]. Чтобы этого избежать, измерения следует проводить при таких значениях [S], которые соответствуют равным интервалам по шкале обратных величин.
Рисунок 86. График преобразования Лайнуивера-Бэрка
Оценки Км имеют практическую ценность. При концентрациях субстрата, в 100 раз превышающих Км, фермент будет работать практически с максимальной скоростью, поэтому максимальная скорость (Vmах) будет отражать количество присутствующего активного фермента. Это немаловажное обстоятельство используют для оценки содержания фермента в препарате. Значение Км позволяет ориентироваться, какое количество субстрата следует добавить для определения Vmax. Графики, построенные в обратных координатах, находят широкое применение при оценке действия ингибиторов.
С другой стороны Км связана с константой диссоциации фермент-субстратного комплекса то есть со сродством фермента к субстрату (Рисунок 87).
Рисунок 87. Соотношение между Км и Кd
Сродство фермента к субстрату равно величине, обратной константе диссоциации К комплекса Enz — S:
Иными словами, чем слабее выражена тенденция фермент-субстратного комплекса к диссоциации, тем выше сродство фермента к субстрату. Мерой Kd может ориентировочно служить значение Км для данного фермента по отношению к его субстрату. Однако это возможно только в том случае, если справедливо допущение, использовавшееся при выводе уравнения Михаэлиса — Ментен. Оно состояло в том, что первая стадия ферментативной реакции идет быстро и при этом всегда на этой стадии поддерживается равновесие. Другими словами, скорость диссоциации Enz — S на Enz + S намного выше, чем скорость диссоциации на фермент — продукт: Из уравнения Михаэлиса — Ментен следует, что величина [S], при которой v = 1/2Vmax, равна
При этих условиях 1/KM =1/Кd и равно сродству фермента к субстрату. Если k2 + k-1 не равно k-1, то 1/Км даст заниженную оценку сродства.
Ограничения, присущие уравнению Михаэлиса-Ментен
Некоторые ферменты и другие лигандсвязывающие белки, например гемоглобин, не подчиняются классической кинетике насыщения Михаэлиса — Ментен. График зависимости v от [S] носит в этом случае сигмоидный характер. Это обычно указывает на кооперативное связывание субстрата многими центрами — связывание с одним центром влияет на связывание с другим, как это происходит с гемоглобином. В таком случае описанный выше метод графической оценки концентрации субстрата, при которой скорость реакции составляет половину максимальной, оказывается непригодным (прямой в соответствующих координатах уже не получается). Здесь следует обратиться к графическому представлению уравнения Хилла, первоначально предложенного для описания кооперативного связывания О2 с гемоглобином.
Рисунок 88. Уравнение Хилла и график, им описываемый
Уравнение Хилла, преобразованное так, чтобы график в соответствующих координатах представлял собой прямую, имеет вид, где ќ — константа. Из уравнения следует, что в условиях, когда [S] мало по сравнению с ќ, скорость реакции возрастает как n-я степень [S]. Приведен график Хилла, построенный по кинетическим данным для фермента, характеризующегося кооперативным связыванием субстрата (Рисунок 88). График зависимости log (V/Vmax — V) от log [S] представляет собой прямую с тангенсом угла наклона, равным n, где п — эмпирический параметр, зависящий от числа субстрат связывающих центров и характера взаимодействия между ними. При п = 1 связывающие центры не зависят друг от друга. При п> 1 между центрами имеется кооперативное взаимодействие; чем больше п, тем выше степень кооперативности и тем более выражена сигмоидность кривых насыщения. При п <1 говорят об отрицательной кооперативности. Когда скорость реакции равна половине максимальной (V = Vmax/2, V/ (Vmax -V) = 1 и log [V/ (Vmax —V)] = 0. Таким образом, чтобы определить величину S50 (концентрацию субстрата, при которой скорость вдвое меньше максимальной), нужно из точки на прямой, для которой log [V/ (Vmax — V)] = 0, опустить перпендикуляр на ось х.
Ингибирование активности ферментов
Ингибиторы — это химические вещества, которые уменьшают скорость ферментативной реакции.
Различают два больших класса ингибиторов ферментативной активности — конкурентные и неконкурентные — на основании того, ослабляется (конкурентное ингибирование) или не ослабляется (неконкурентное ингибирование) их ингибирующее действие при повышении концентрации субстрата. На практике многие ингибиторы не проявляют тех свойств, которые характерны для чисто конкурентного или чисто неконкурентного ингибирования. Другой способ классификации ингибиторов основывается на характере места их связывания. Одни из них связываются с ферментом в том же месте, что и субстрат (в каталитическом центре), а другие — на значительном расстоянии от активного центра (в аллостерическом центре). Так же ингибирование подразделятся
Конкурентное ингибирование аналогами субстрата
Классическое конкурентное ингибирование основано на связывании ингибитора с субстрат-связывающим (каталитическим) центром. Химическая структура аналога субстрата, действующего как ингибитор (I), обычно сходна со структурой субстрата (S). Поэтому ингибитор может обратимо связываться с ферментом, образуя вместо Enz — S комплекс Enz — I, т. е. фермент-ингибиторный комплекс. Когда в реакционной смеси одновременно присутствуют и субстрат, и. ингибитор указанного типа, они конкурируют за один и тот же связывающий центр на поверхности фермента. Один из наиболее подробно изученных примеров конкурентного ингибирования — это ингибирование сукцинатдегидрогеназы малонатом (I), конкурирующим за один и тот же центр с субстратом сукцинатом (S). В случае конкурентного ингибирования идет воздействие на Км, при повышении концентрации субстрата, вероятность связывания с ингибитором уменьшается и скорость реакции увеличивается, таким образом при конкурентном ингибировании скорость реакции может быть близкой к максимальной в норме, но при больших концентрациях субстрата.
Обратимое неконкурентное ингибирование
Как следует уже из самого названия, в этом случае конкуренция между субстратом (S) и ингибитором (I) отсутствует. При этом ингибитор обычно ничем не напоминает S и, как можно предположить, связывается с другим участком фермента. Обратимые неконкурентные ингибиторы понижают максимальную скорость, достижимую при данном количестве фермента (понижают Vmax),но, как правило, не влияют на Км. Поскольку I и S связываются с разными центрами, возможно образование как комплекса Enz — I, так и комплекса Enz — IS. Комплекс Enz — IS тоже распадается с образованием продукта, однако с меньшей скоростью, чем Enz — S; поэтому реакция будет замедляться, но не остановится. Таким образом, могут протекать следующие конкурентные реакции:
Необратимое неконкурентное ингибирование
Ферментативная активность может уменьшаться в присутствии многих «ядов», таких, как иодацетамид, ионы тяжелых металлов (Ag+, Hg2+) окисляющие агенты и т. д. В присутствии одного или нескольких субстратов или продуктов скорость инактивации фермента может снижаться. В данном случае ингибирование происходит за счет частичной денатурации фермента.
Способы регуляции работы ферментов in vivo
Клетка также регулирует скорость ферментативных реакций, для контроля своего метаболизма. Но клетка не может изменять температуру и рН своей среды, а изменение концентрации субстратов является сигналом для изменения скорости реакций. Такая ситуация связна с понятием гомеостаза. Гомеостаз — это состояние постоянных состава внутренней среды клетки и организма и их физиологических функций (температура и рН).
Поэтому в живой клетке скорость ферментативных реакций можно регулировать, изменяя следующие параметры:
1) абсолютное количество присутствующего фермента;
2) пул реагентов (помимо фермента);
3) каталитическую эффективность фермента.
Большинство форм жизни использует все три типа регуляции.
Регуляция количества фермента путем регуляции скорости его синтеза и распада
Абсолютное количество фермента в клетке определяется скоростями его синтеза (Kсинт) и распада (Kрасп). Соответственно количество фермента увеличивается либо в результате повышения скорости его синтеза (увеличением Kсинт), либо снижения скорости распада (уменьшением Kрасп), либо обоими способами сразу. Регуляция осуществляется на уровне экспрессии генов. Увеличивая скорость экспрессии генов прежде всего транскрипции и в меньшей степени трансляции можно увеличивать количество белка. Блокируя транскрипцию генов можно уменьшать до полного исчезновения количество закодированного в гене белка.
Превращение проферментов в активные ферменты
Ферментативная активность может регулироваться путем превращения неактивного профермента в каталитически активную форму. Чтобы перейти в такую форму, профермент должен подвергнуться ограниченному протеолизу, сопровождающемуся конформационными изменениями; при этом происходит либо демаскирование каталитического центра, либо его формирование. Синтез в форме каталитически неактивных проферментов является характерным свойством пищеварительных ферментов, а также ферментов системы свертывания крови и системы фибринолиза.
Компартментация ферментов
Роль компартментации (пространственного разделения) метаболических процессов в клетках эукариот, в том числе в клетках млекопитающих, трудно переоценить. Локализация специфических метаболических процессов в цитозоле или в клеточных органеллах облегчает независимую регуляцию этих процессов. Развитая компартментация метаболических процессов особенно характерна для высших форм живых организмов — она позволяет осуществлять наиболее тонкую регуляцию метаболизма. Одновременно при этом возникает новая проблема — прохождение метаболитов через разделяющие барьеры. Эта проблема решается с помощью «челночных механизмов», переводящих метаболит в форму, которая способна проходить через барьер. Затем по другую сторону барьера происходит обратное превращение метаболита в первоначальную форму. В связи с наличием барьеров возникает потребность в функционировании, например, цитозольной и митохондриальной форм некоторых ферментов. Поскольку эти формы фермента физически разделены, их независимая регуляция облегчается.
Аллостерическая регуляция
В случае аллостерической регуляции низко-молекулярные соединения или аллостерические регуляторы связываются с ферментом в результате происходит изменение конформации белка. Необходимо вспомнить что при при встраивании этой молекулы изменяются связи в составе белка, что характерно для связывания белка с любой молекулой в результате происходит перераспределение связей и как результат изменение конформации белка, эти молекулы могут привести к двум противоположным результатам. В зависимости от молекулы связи могут различаться, а, следовательно, и перестройки в молекуле белка, поэтому аллостеричекие модуляторы могут изменять конформацию белка либо в направлении увеличения сродства к субстрату, следовательно, увеличению активности белка, тогда молекула является положительным модулятором. И наоборот молекула, встраиваясь, изменяет конформацию белка, и в результате конформация белка становится менее активной, то есть происходит неконкурентное обратимое ингибирование.
Примерно в 1963 г. Моно обратил внимание на отсутствие структурного сходства между ингибитором, действующим на фермент по принципу обратной связи, и субстратом этого фермента. Отсутствие изостеричности с субстратом позволяет говорить об аллостеричности соответствующих эффекторов. Исходя из этого, Моно предположил, что ферменты, регулируемые такими аллостерическими эффекторами (в частности, ингибиторами по принципу обратной связи), связывают эффектор в аллостерическом центре, физически не совпадающем с каталитическим центром. Таким образом, аллостерические ферменты — это ферменты, активность каталитического центра которых изменяется под действием аллостерических эффекторов, связывающихся в аллостерическом центре. Данные о наличии у регуляторных ферментов физически обособленных аллостерических центров сводятся к следующему.
1.Регуляторные ферменты после модификации химическими или физическими методами часто становятся нечувствительны к аллостерическим эффекторам, сохраняя каталитическую активность.
2.Аллостерические эффекторы часто защищают каталитический центр от денатурации в условиях, когда субстрат такого защитного действия не оказывает.
3.Обнаружены мутантные клетки бактерий и млекопитающих, в которых регуляторные ферменты имели существенно иные регуляторные свойства, чем ферменты из клеток дикого типа, но обладали в точности такими же каталитическими свойствами.
4.Показано, что связывание субстратов и аллстерических эффекторов с регуляторным ферментом происходит независимо.
5.В некоторых ферментах (например, в АТКазе) аллостерический и каталитический центры локализованы в разных протеомерах.
Ковалентная модификация ферментов
Обратимое изменение каталитической активности ферментов может осуществляться путем ковалентного присоединения фосфатной группы (преобладает у млекопитающих) или нуклеотида (преобладает у бактерий). Ферменты, подверженные ковалентной модификации, которая сопровождается изменением их активности, называют обратимо модифицируемыми ферментами.
Обратимо модифицируемые ферменты могут находиться в двух состояниях, одно из которых характеризуется высокой, а другое — низкой каталитической эффективностью. В зависимости от конкретного случая более активным катализатором может быть либо фосфо-, либо дефосфофермент.
Обычно фосфорилируется специфический остаток серина и образуется остаток О-фосфосерина; реже фосфорилируется остаток тирозина с образованием остатка О-фосфотирозина. Хотя обратимо модифицируемый фермент может содержать много остатков серина или тирозина, фосфорилирование происходит в высшей степени избирательно и затрагивает лишь небольшое число (1—3) остатков. Эти участки, по всей вероятности, не являются частью каталитического центра; мы, таким образом, имеем еще один пример аллостерических эффектов.
Для модификации необходимы молекула — донор модифицирующей группировки и фермент, осуществляющий модификацию. У эукариот донором фосфатной группы для модификации является АТФ, модификацию осуществляют ферменты протеин киназы. Протеинкиназы — это очень большой класс белков, специфично фосфорилирующих различные ферменты. У прокариот донором АДФ-рибозы является NAD. Различие модифицирующих групп повлекло за собой расхождение модифицирующих и демодифицирующих ферментов. По этому у эукариот нет ферментов снимающих АДФ-рибозу с модифицированного фермента. Эта особенность стала основой для действия некоторых токсинов. Например дифтерийный токсин АДФ-рибозилирует рибосому, полностью блокируя ее функции, у клетки человека нет ферментов, удаляющих модифицирующую группу, а следовательно, клетка погибает без возможности запустить биосинтез белка. Холерный токсин необратимо АДФ-рибозилирует аденилатциклазу в цитоплазматической мембране клеток эпителия тонкого кишечника, в результате все процессы всасывания полностью нарушаются.
Изоферменты
Изоферменты — это ферменты отличающиеся активностью у одного организма в разных тканях или в ходе онтогенеза а так же у разных особей одного вида.
Изоферменты отличаются как распределением в популяции, так в тканях организма. Также как и изобелки изоферменты могут по разному распределяться как в тканях организма, так и в ходе онтогенеза. Термин «изофермент» («изоэнзим») охватывает все вышеупомянутые физически различимые белки с данной каталитической активностью, однако на практике, и особенно в клинической медицине, его употребляют в более узком смысле, подразумевая физически различимые и поддающиеся разделению формы данного фермента, присутствующие в различных типах клеток данного эукариотического организма, например человека. Изоэнзимы неизменно обнаруживаются в сыворотке и в тканях всех позвоночных, насекомых и в одноклеточных организмах. При этом число ферментов и их содержание сильно варьируют. Известны изоферментные формы дегидрогеназ, оксидаз, трансаминаз, фосфатаз, трансфосфорилаз и протеолитических ферментов. В различных тканях могут находиться разные изоферменты, и эти изоферменты могут иметь неодинаковое сродство к субстратам.
Изоэнзимы лактатдегидрогеназы различаются на уровне четвертичной структуры. Олигомерная молекула лактатдегидрогеназы (мол. масса 130000) состоит из четырех протомеров двух типов, Н и М (оба с мол. массой около 34000). Каталитической активностью обладает только тетрамерная молекула. Если порядок соединения протомеров не имеет значения, то протомеры могут быть скомпонованы пятью способами:
НННН
НННМ
ННММ
НМММ
ММММ
Маркерт подобрал условия для разрушения и реконструкции четвертичной структуры и сумел выяснить взаимоотношения между изоэнзимами лактатдегидрогеназы. Расщепление и реконструкция лактатдегидрогеназ I1 и I5 не приводят к образованию новых изоэнзимов. Следовательно, эти два изоэнзима содержат только один тип протомеров. Когда такой же процедуре была подвергнута смесь лактатдегидрогеназ I1 и I5, появились также формы I2, I3 и I4. Соотношение изозимов соответствует приведенному ниже субъединичному составу:
I1— НННН
I2— НННМ
I3-ННММ
I4— НМММ
I5 -ММММ
Синтез Н- и М-субъединиц детерминируется разными генетическими локусами, и они по-разному экспрессируются в разных тканях (например, в сердечной и скелетной мышцах).
Также примером могут быть изоформы цитохрома Р450, существующего в двух изоформах нормальной и высокоактивной, которая является причиной предрасположенности организма к различным онкозаболеваниям. Чаще всего причиной возникновения изоформ белков являются точечные мутации, не подвергшиеся давлению естественного отбора.
Тема: «СВОЙСТВА И КЛАССИФИКАЦИЯ ФЕРМЕНТОВ. ВЛИЯНИЕ ТЕМПЕРАТУРЫ И рН СРЕДЫ НА АКТИВНОСТЬ ФЕРМЕНТОВ. СПЕЦИФИЧНОСТЬ ДЕЙСТВИЯ ФЕРМЕНТОВ. ОПРЕДЕЛЕНИЕ АКТИВНОСТИ ФЕРМЕНТОВ»
1. Химическая природа ферментов. Значение ферментов для жизнедеятельности организма.
2. Основные свойства ферментов. Влияние концентрации фермента и субстрата, температуры и рН среды на скорость ферментативной реакции. Олигодинамичность и обратимость действия ферментов.
3. Специфичность действия ферментов (абсолютная, относительная и стереохимическая). Примеры.
4. Важнейший признак, положенный в основу классификации ферментов. Понятие о кодовом номере фермента. Классы ферментов: оксидоредуктазы, трансферазы, гидролазы, лиазы, изомеразы, лигазы. Тип и общее уравнение катализируемых реакций, принципы формирования подклассов.
5. Номенклатура ферментов (понятие о систематическом и рабочем (рекомендуемом) названиях ферментов, их использование).
6. Определение активности ферментов. Аналитические методы, применяемые для определения активности. Единицы общей, удельной, молекулярной активности ферментов, их использование. Формула для расчёта общей активности фермента в сыворотке крови.
| Раздел 7.1 | Химическая природа ферментов. Значение ферментов для жизнедеятельности организма. |
| | |
| 7.1.1. Протекание процессов обмена веществ в организме определяется действием многочисленных ферментов — биологических катализаторов белковой природы. Они ускоряют химические реакции и сами при этом не расходуются. Термин «фермент» происходит от латинского слова fermentum — закваска. Наряду с этим понятием в литературе используется равноценный термин «энзим» (en zyme — в дрожжах) греческого происхождения. Отсюда раздел биохимии, изучающий ферменты, получил название «энзимология». Энзимология составляет основу познания на молекулярном уровне важнейших проблем физиологии и патологии человека. Переваривание пищевых веществ и их использование для выработки энергии, образование структурных и функциональных компонентов тканей, сокращение мышц, передача электрических сигналов по нервным волокнам, восприятие света глазом, свертывание крови — каждый из этих физиологических механизмов имеет в основе каталитическое действие определенных ферментов. Было показано, что многочисленные заболевания непосредственно нарушением ферментативного катализа; определение активности ферментов в крови и других тканях даёт ценные сведения для медицинской диагностики; ферменты или их ингибиторы могут применяться как лекарственные вещества. Таким образом, знание важнейших особенностей ферментов и катализируемых ими реакций необходимо при рациональном подходе к изучению заболеваний человека, их диагностике и лечению. 7.1.2. Вещества, превращения которых катализируют ферменты, называются субстратами. Фермент, соединяясь с субстратом, образует фермент-субстратный комплекс (рисунок 7.1).
Рисунок 7.1. Образование фермент-субстратного комплекса в ходе катализируемой реакции. Образование этого комплекса способствует снижению энергетического барьера, который нужно преодолеть молекуле субстрата для вступления в реакцию (рисунок 7.2). По завершении реакции фермент-субстратный комплекс распадается на продукт (продукты) и фермент. Фермент по окончании реакции возвращается в своё исходное состояние и может взаимодействовать с новой молекулой субстрата.
Рисунок 7.2. Влияние фермента на энергетический барьер реакции. Ферменты, действуя как катализаторы, снижают энергию активации, которая требуется для того, чтобы могла произойти реакция. 7.1.3. Для ферментов характерны свойства, присущие всем белкам. В частности, молекулы ферментов, как и других белков, построены из α-аминокислот, соединённых пептидными связями. Поэтому растворы ферментов дают положительную биуретовую реакцию, а их гидролизаты — положительную нингидриновую реакцию. Нативные свойства и функции ферментов определяются наличием определённой пространственной структуры (конформации) их полипептидной цепи. Изменение этой структуры в результате тепловой денатурации приводит к потере каталитических свойств. Наличие у ферментов высокой молекулярной массы обусловливает их неспособность к диализу, а присутствие в молекулах заряженных функциональных групп — подвижность в электрическом поле. Как и другие белки, ферменты образуют коллоидные растворы, из которых могут осаждаться ацетоном, спиртом, сульфатом аммония — веществами, способствующими разрушению гидратной оболочки и нейтрализации электрического заряда. |

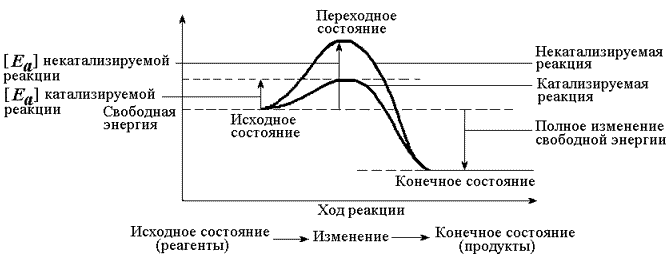
| Раздел 7.2 | Основные свойства ферментов. Олигодинамичность и обратимость действия ферментов. Влияние концентрации фермента и субстрата, температуры и рН среды на скорость ферментативной реакции. |
| | |
| 7.2.1. Белковая природа ферментов обусловливает появление у них ряда свойств, в целом нехарактерных для неорганических катализаторов: олигодинамичность, специфичность, зависимость скорости реакции от температуры, рН среды, концентрации фермента и субстрата, присутствия активаторов и ингибиторов. Под олигодинамичностью ферментов понимают высокую эффективность действия в очень малых количествах. Такая высокая эффективность объясняется тем, что молекулы ферментов в процессе своей каталитической деятельности непрерывно регенерируют. Типичная молекула фермента может регенерировать миллионы раз в минуту. Надо сказать, что и неорганические катализаторы также способны ускорять превращение такого количества веществ, которое во много раз превышает их собственную массу. Но ни один неорганический катализатор не может сравниться с ферментами по эффективности действия. Примером может служить фермент реннин, вырабатываемый слизистой оболочкой желудка жвачных животных. Одна молекула его за 10 минут при 37°С способна вызывать коагуляцию (створаживание) порядка миллиона молекул казеиногена молока. Другой пример высокой эффективности ферментов даёт каталаза. Одна молекула этого фермента при 0°С расщепляет за секунду около 50 000 молекул пероксида водорода: 2 Н2О2 Действие каталазы на пероксид водорода заключается в изменении величины энергии активации этой реакции приблизительно от 75 кДж/моль без катализатора до 21 кДж/моль в присутствии фермента. Если же в качестве катализатора этой реакции используется коллоидная платина, то энергия активации составляет всего 50 кДж/моль. 7.2.2. При изучении влияния какого-либо фактора на скорость ферментативной реакции все прочие факторы должны оставаться неизменными и по возможности иметь оптимальное значение. Мерой скорости ферментативных реакций служит количество субстрата, подвергшегося превращению в единицу времени, или количество образовавшегося продукта. Изменение скорости проводят на начальной стадии реакции, когда продукт ещё практически отсутствует, и обратная реакция не идёт. Кроме того, на начальной стадии реакции концентрация субстрата соответствует его исходному количеству. 7.2.3. Зависимость скорости ферментативной реакции (V) от концентрации фермента [Е] (рисунок 7.3). При высокой концентрации субстрата (многократно превышающей концентрацию фермента) и при постоянстве других факторовскорость ферментативной реакции пропорциональна концентрации фермента. Поэтому зная скорость реакции, катализируемой ферментом, можно сделать вывод о его количестве в исследуемом материале.
Рисунок 7.3. Зависимость скорости ферментативной реакции от концентрации фермента 7.2.4. Зависимость скорости реакции от концентрации субстрата [S]. График зависимости имеет вид гиперболы (рисунок 7.4). При постоянной концентрации фермента скорость катализируемой реакции возрастает с увеличением концентрации субстрата до максимальной величины Vmax, после чего остаётся постоянной. Это следует объяснить тем, что при высоких концентрациях субстрата все активные центры молекул фермента оказываются связанными с молекулами субстрата. Любое избыточное количество субстрата может соединиться с ферментом лишь после того, как образуется продукт реакции и освободится активный центр.
Рисунок 7.4. Зависимость скорости ферментативной реакции от концентрации субстрата. Зависимость скорости реакции от концентрации субстрата может быть выражена уравнением Михаэлиса — Ментен:
где V — скорость реакции при концентрации субстрата [S] , Vmax —максимальная скорость и KM —константа Михаэлиса. Константа Михаэлиса равна концентрации субстрата, при которой скорость реакции составляет половину максимальной. Определение KM и Vmax имеет важное практическое значение, так как позволяет количественно описать большинство ферментативных реакций, включая реакции с участием двух и более субстратов. Различные химические вещества, изменяющие активность ферментов, по-разному воздействуют на величины Vmax и KM. 7.2.5. Зависимость скорости реакции от t – температуры, при которой протекает реакция (рисунок 7.5), имеет сложный характер. Значение температуры, при котором скорость реакции максимальна, представляет собой температурный оптимум фермента. Температурный оптимум большинства ферментов организма человека приблизительно равен 40°С. Для большинства ферментов оптимальная температура равна или выше тойц температуры, при которой находятся клетки.
Рисунок 7.5. Зависимость скорости ферментативной реакции от температуры. При более низких температурах (0° — 40°С) скорость реакции увеличивается с ростом температуры. При повышении температуры на 10°С скорость ферментативной реакции удваивается (температурный коэффициент Q10 равен 2). Повышение скорости реакции объясняется увеличением кинетической энергии молекул. При дальнейшем повышении температуры происходит разрыв связей, поддерживающих вторичную и третичную структуру фермента, то есть тепловая денатурация. Это сопровождается постепенной потерей каталитической активности. 7.2.6. Зависимость скорости реакции от рН среды (рисунок 7.6). При постоянной температуре фермент работает наиболее эффективно в узком интервале рН. Значение рН, при котором скорость реакции максимальна, представляет собой оптимум рН фермента. У большинства ферментов организма человека оптимум рН находится в пределах рН 6 – 8, но есть ферменты, которые активны при значениях рН, лежащих за пределами этого интервала (например, пепсин, наиболее активный при рН 1,5 — 2,5). Изменение рН как в кислую, так и в щелочную сторону от оптимума приводит к изменению степени ионизации кислых и основных групп аминокислот, входящих в состав фермента (например, СООН-группы аспартата и глутамата, NН2-группы лизина и т.д.). Это вызывает изменение конформации фермента, в результате чего изменяется пространственная структура активного центра и снижение его сродства к субстрату. Кроме того, при экстремальных значениях рН происходит денатурация фермента и его инактивация.
Рисунок 7.6. Зависимость скорости ферментативной реакции от рН среды. Следует отметить, что свойственный ферменту оптимум рН не всегда совпадает с рН его непосредственного внутриклеточного окружения. Это позволяет предположить, что среда, в которой находится фермент, в какой-то мере регулирует его активность. 7.2.7. Зависимость скорости реакции от присутствия активаторов и ингибиторов. Активаторы повышают скорость ферментативной реакции. Ингибиторы понижают скорость ферментативной реакции. В качестве активаторов ферментов могут выступать неорганические ионы. Предполагают, что эти ионы заставляют молекулы фермента или субстрата принять конформацию, способствующую образованию фермент-субстратного комплекса. Тем самым увеличивается вероятность взаимодействия фермента и субстрата, а следовательно и скорость реакции, катализируемой ферментом. Так, например, активность амилазы слюны повышается в присутствии хлорид-ионов. |
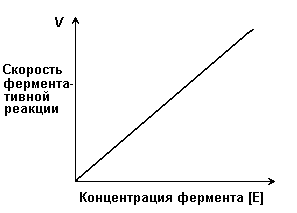
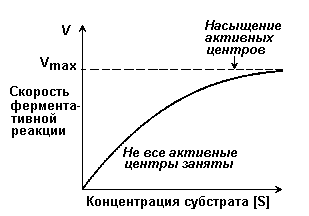
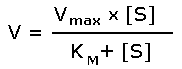 ,
,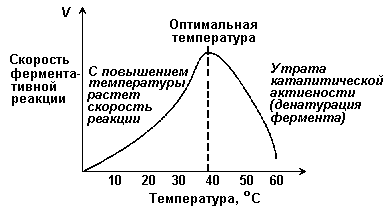
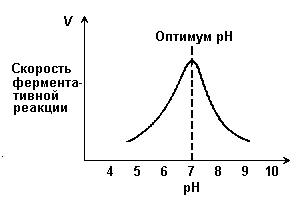
| Раздел 7.3 | Специфичность действия ферментов (абсолютная, относительная и стереохимическая). |
| | |
| 7.3.1. Важным свойством, отличающим ферменты от неорганических катализаторов, является специфичность действия. Как известно, структура активного центра фермента комплементарна структуре его субстрата. Поэтому фермент из всех имеющихся в клетке веществ выбирает и присоединяет только свой субстрат. Для ферментов характерна специфичность не только по отношению к субстрату, но и в отношении пути превращения субстрата. У ферментов различают абсолютную, относительную и стереохимическую специфичность. 7.3.2. Абсолютная специфичность – избирательная способность фермента катализировать только единственное из возможных превращений одного субстрата. Это можно объяснить конформационной и электростатической комплементарностью молекул субстрата и фермента. Например, фермент аргиназа катализирует только гидролиз аминокислоты аргинина, фермент уреаза – только расщепление мочевины и не действуют на другие субстраты. 7.3.3. Относительная специфичность – избирательная способность фермента катализировать однотипные превращения сходных по строению субстратов. Такие ферменты оказывают воздействие на одинаковые функциональные группы или на один и тот же тип связей в молекулах субстратов. Так, например, разные гидролитические ферменты действуют на определённый тип связей:
Действие этих ферментов распространяется на большое число субстратов, что позволяет организму обойтись малым количеством пищеварительных ферментов — иначе их потребовалось бы намного больше. 7.3.4. Стереохимическая (оптическая) специфичность — избирательная способность фермента катализировать превращение только одного из возможных пространственных изомеров субстрата. Так, большинство ферментов млекопитающих катализирует превращение толькл L-изомеров аминокислот, но не D-изомеров. ферменты, участвующие в обмене моносахаридов, наоборот, катализируют превращение только D-, но не L-фосфосахаров. Гликозидазы специфичны не только к моносахаридному фрагменту, но и характеру гликозидной связи. Например, α-амилаза расщепляет α–1,4-гликозидные связи в молекуле крахмала, но не действует на α–1,2-гликозидные связи в молекуле сахарозы. |
| Раздел 7.4 | Основные принципы, положенные в основу современной классификации и номенклатуры ферментов. |
| | |
| 7.4.1. В настоящее время известно более двух тысяч химических реакций, катализируемых ферментами, и число это непрерывно возрастает. Чтобы ориентироваться в таком множестве превращений. возникла настоятельная необходимость в систематизированной классификации и номенклатуре, при помощи которой любой фермент можно было бы точно идентифицировать. Номенклатура, которой пользовались до середины XX века, была весьма далека от совершенства. Исследователи, открывая новый фермент, давали ему название по своему усмотрению, что неизбежно вело к путанице и всевозможным противоречиям. Некоторые названия оказались ошибочными, другие ничего не говорили о природе катализируемой реакции. Учёные разных школ часто употребляли разные названия для одного и того же фермента или, наоборот, одно и то же название для нескольких разных ферментов. Было решено разработать рациональную международную классификацию и номенклатуру ферментов, которой могли бы пользоваться биохимики всех стран. С этой целью при Международном союзе биохимии и молекулярной биологии (International Union of Biochemistry and Molecular Biology, IUВMB) была создана Комиссия по ферментам, предложившая в 1964 году основные принципы такой классификации и номенклатуры. Она постоянно совершенствуется и дополняется, в настоящее время действует уже шестая редакция этой номенклатуры (1992 год), к которой ежегодно выходят дополнения. |
7.4.2. В основу классификации положен важнейший признак, по которому один фермент отличается от другого — это катализируемая им реакция. Число типов химических реакций сравнительно невелико, что позволило разделить все известные в настоящее время ферменты на 6 важнейших классов, в зависимости от типа катализируемой реакции. Такими классами являются:
- оксидоредуктазы (окислительно-восстановительные реакции);
- трансферазы (перенос функциональных групп);
- гидролазы (реакции расщепления с участием воды);
- лиазы (разрыв связей без участия воды);
- изомеразы (изомерные превращения);
- лигазы (синтез с затратой молекул АТФ).
7.4.3. Ферменты каждого класса делят на подклассы, руководствуясь строением субстратов. В подклассы объединяют ферменты, действующие на сходно построенные субстраты. Подклассы разбивают на подподклассы, в которых ещё строже уточняют структуру химических групп, отличающих субстраты друг от друга. Внутри подподклассов перечисляют индивидуальные ферменты. Все подразделения классификации имеют свои номера. Таким образом, любой фермент получает свой уникальный кодовый номер, состоящий из четырёх чисел, разделённых точками. Первое число обозначает класс, второе — подкласс, третье — подподкласс, четвёртое — номер фермента в пределах подподкласса. Например, фермент α-амилаза, расщепляющая крахмал, обозначается как 3.2.1.1, где:
3 — тип реакции (гидролиз);
2 — тип связи в субстрате (гликозидная);
1 — разновидность связи (О-гликозидная);
1 — номер фермента в подподклассе
Вышеописанный десятичный способ нумерации имеет одно важное преимущество: он позволяет обойти главное неудобство сквозной нумерации ферментов, а именно: необходимость при включении в список вновь открытого фермента изменять номера всех последующих. Новый фермент может быть помещён в конце соответствующего подподкласса без нарушения всей остальной нумерации. Точно так же при выделении новых классов, подклассов и подподклассов их можно добавлять без нарушения порядка нумерации ранее установленных подразделений. Если после получения новой информации возникает необходимость изменить номера некоторым ферментам, прежние номера не присваивают новым ферментам, чтобы избежать недоразумений.
Говоря о классификации ферментов, следует также отметить, что ферменты классифицируются не как индивидуальные вещества, а как катализаторы определённых химических превращений. Ферменты, выделенные из разных биологических источников и катализирующие идентичные реакции, могут существенно отличаться по своей первичной структуре. Тем не менее в классификационном списке все они фигурируют под одним и тем же кодовым номером.
Итак, знание кодового номера фермента позволяет:
- устранить неоднозначности, если разные исследователи используют одно и то же название для различных ферментов;
- сделать поиск информации в литературных базах данных более эффективным;
- получить в других базах данных дополнительную информацию о последовательности аминокислот, пространственной структуре фермента, генах, кодирующих ферментные белки.
аздел 7.5
Понятие о систематическом и рабочем названии фермента, их использование.
7.5.1. Система классификации, разработанная Комиссией по ферментам, включает также и вновь созданную номенклатуру ферментов, которая строится по специальным принципам. Согласно рекомендациям IUBMB, ферменты получают два рода названий: систематическое и рабочее (рекомендуемое).
7.5.2. Систематическое название составляется из двух частей. Первая часть содержит название субстрата или субстратов, часто — наименование кофермента, вторая часть указывает на природу катализируемой реакции и включает название класса, к которому относится данный фермент. При необходимости приводится дополнительная информация о реакции в скобках после второй части названия. Систематическое название присваивается только тем ферментам, каталитическое действие которых полностью изучено.
Например, систематическое название α-амилазы — 1,4-α-D-глюкан-глюканогидролаза. Конечно, такое название очень неудобно для запоминания и произнесения. Поэтому наряду с систематическими Комиссия по ферментам IUBMB даёт рекомендует использовать рабочие (упрощённые) названия ферментов.
7.5.3. Рабочее название фермента должно быть достаточно коротким для употребления. В качестве рабочего названия в ряде случаев может быть использовано тривиальное название, если оно не является ошибочным или двусмысленным. В других случаях оно строится на тех же общих принципах, что и систематическое название, но с минимальной детализацией. Конкретные примеры систематических и рабочих названий ферментов приводятся в следующем разделе данной темы курса. В научных публикациях при первом упоминании о ферменте принято указывать его систематическое название и кодовый номер, а в дальнейшем пользоваться его рабочим названием.
7.5.4. Основные правила построения систематических и рабочих названий разных классов ферментов:
Оксидоредуктазы
 Систематическое название ферментов этого класса строится по схеме донор: акцептор — оксидоредуктаза.Согласно тривиальной номенклатуре, оксидоредуктазы, отщепляющие атомы водорода или электроны и переносящие их на любой акцептор, кроме кислорода, называются дегидрогеназами. Оксидоредуктазы, использующие кислород в качестве акцептора атомов водорода или электронов, называются оксидазами.Некоторые ферменты, которым свойственно преимущественно восстанавливающее действие, носят названиередуктаз. Все перечисленные наименования могут быть использованы для построения рабочего названияоксидоредуктаз.
Систематическое название ферментов этого класса строится по схеме донор: акцептор — оксидоредуктаза.Согласно тривиальной номенклатуре, оксидоредуктазы, отщепляющие атомы водорода или электроны и переносящие их на любой акцептор, кроме кислорода, называются дегидрогеназами. Оксидоредуктазы, использующие кислород в качестве акцептора атомов водорода или электронов, называются оксидазами.Некоторые ферменты, которым свойственно преимущественно восстанавливающее действие, носят названиередуктаз. Все перечисленные наименования могут быть использованы для построения рабочего названияоксидоредуктаз.
Трансферазы
Систематическое название ферментов, ускоряющих такие реакции, составляют по форме донор:акцептор (транспортируемая группа) трансфераза. В рабочем названии обычно указывается только один специфический субстрат или продукт наряду с названием транспортируемой группировки.
Гидролазы
 Систематическое название составляется по форме субстрат-гидролаза. У гидролаз, специфически отщепляющих определённую группу, эта группа может быть указана в виде префикса. Рабочее название чаще всего составляется из названия гидролизуемого субстрата с добавлением окончания -аза. Следует, однако, отметить, что вследствие достаточно сложного и зачастую до конца не выявленного характера специфичности многих гидролаз не всегда удаётся дать им систематическое название. В этих случаях рекомендовано использовать эмпирические названия, присвоенные им при первом описании. Так, не имеют систематического названия такие ферменты, как пепсин, папаин, тромбин.
Систематическое название составляется по форме субстрат-гидролаза. У гидролаз, специфически отщепляющих определённую группу, эта группа может быть указана в виде префикса. Рабочее название чаще всего составляется из названия гидролизуемого субстрата с добавлением окончания -аза. Следует, однако, отметить, что вследствие достаточно сложного и зачастую до конца не выявленного характера специфичности многих гидролаз не всегда удаётся дать им систематическое название. В этих случаях рекомендовано использовать эмпирические названия, присвоенные им при первом описании. Так, не имеют систематического названия такие ферменты, как пепсин, папаин, тромбин.
Лиазы
 Систематическое название ферментов строится по схеме: субстрат-отщепляемая группа-лиаза. Чтобы уточнить, какая группа отщепляется, используются префиксы «карбокси-«, «аммиак», «гидро-» и т.д. В качестве рабочих названий ферментов сохраняются тривиальные названия типа «декарбоксилаза», «альдолаза», «дегидратаза», «десульфгидраза». Лиазы делятся на подклассы в зависимости от характера разрываемых связей
Систематическое название ферментов строится по схеме: субстрат-отщепляемая группа-лиаза. Чтобы уточнить, какая группа отщепляется, используются префиксы «карбокси-«, «аммиак», «гидро-» и т.д. В качестве рабочих названий ферментов сохраняются тривиальные названия типа «декарбоксилаза», «альдолаза», «дегидратаза», «десульфгидраза». Лиазы делятся на подклассы в зависимости от характера разрываемых связей
Изомеразы
Систематическое название ферментов включает название субстрата и слово изомераза, которому предшествует указание типа реакции изомеризации. Рабочие названия подобны (с некоторыми упрощениями) систематическим названиям.
Лигазы
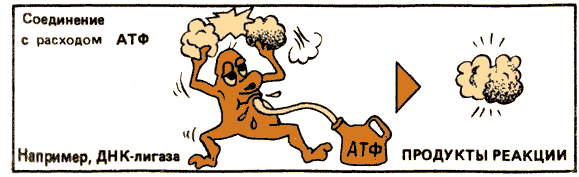 Систематическое название образуется из названий соединяемых субстратов в сочетании со словомлигаза. В скобках указывается продукт, образующийся в результате гидролиза нуклеозидтрифосфата (например, АДФ или АМФ). Рабочее название ферментов этого класса составляется, как правило, из названия продукта реакции в сочетании со словом синтетаза.
Систематическое название образуется из названий соединяемых субстратов в сочетании со словомлигаза. В скобках указывается продукт, образующийся в результате гидролиза нуклеозидтрифосфата (например, АДФ или АМФ). Рабочее название ферментов этого класса составляется, как правило, из названия продукта реакции в сочетании со словом синтетаза.
Рекомендация. Знакомясь в последующем с различными ферментативными реакциями, всегда анализируйте сущность изменений, происходящих в субстратах, и пытайтесь определить по крайней мере класс фермента, катализирующего реакцию. Анализируйте также названия ферментов и соотносите их с процессами, происходящими в реакциях. Это облегчит запоминание названий ферментов и катализируемых ими превращение и позволит больше времени уделить уяснению биологической роли изучаемых процессов.
| Раздел 7.6.1 | ОКСИДОРЕДУКТАЗЫ. |
| | |
| К классу оксидоредуктаз относят ферменты, катализирующие окислительно-восстановительные реакции. Общая схема их может быть представлена следующим образом: AH2+B где AH2 —донор водорода, B — акцептор водорода. В живых организмах окисление осуществляется преимущественно путём отщепления атомов водорода или электронов от субстратов-доноров. Акцепторами атомов водорода или электронов могут быть различные вещества — коферменты (НАД, НАДФ, ФАД, ФМН, глутатион, липоевая кислота, убихинон), цитохромы. железосерные белки и кислород. Подклассы оксидоредуктаз формируются в зависимости от природы функциональной группы донора водорода (электронов). Всего выделяют 19 подклассов. Основными из них являются следующие: Оксидоредуктазы, действующие на СН-ОН-группу доноров. Ферменты, относящиеся к этому подклассу, окисляют спиртовые группы до альдегидных или кетонных групп. В качестве примера можно привести фермент алкогольдегидрогеназу (алкоголь:НАД-оксидоредуктаза; КФ 1.1.1.1). участвующую в метаболизме этанола в тканях:
Кроме окисления спиртов, ферменты этого подкласса участвуют в дегидрировании оксикислот (молочной, яблочной, изолимонной), моносахаридов и других соединений, содержащих гидроксильные группы. Оксидоредуктазы, действующие на альдегидную или кетонную группу доноров. Эти ферменты окисляют альдегиды и кетоны до карбоновых кислот. К примеру, представитель данного подкласса — глицеральдегид-3-фосфатдегидрогеназа (D-глицеральдегид-3-фосфат:НАД-оксидоредуктаза (фосфорилирующая), КФ 1.2.1.12) — катализирует одну из промежуточных реакций распада глюкозы:
Важно отметить, что продукт этой реакции содержит богатую энергией фосфатную связь в 1-ом положении. Остаток фосфорной кислоты, образующий эту связь, может быть перенесён от 1,3-дифосфоглицерата на АДФ с образованием АТФ (см. далее). Оксидоредуктазы, действующие на СН-СН-группу доноров. В результате катализируемых ими реакций СН-СН-группы превращаются в С=С-группы. то есть происходит образование ненасыщенных соединений из насыщенных. Например, фермент цикла трикарбоновых кислот сукцинатдегидрогеназа (сукцинат:акцептор — оксидоредуктаза, КФ 1.3.99.1) ускоряет окисление янтарной кислоты с образованием ненасыщенной фумаровой кислоты:
Оксидоредуктазы, действующие на CH-NH2-группу доноров. Эти ферменты катализируют окислительное дезаминирование аминокислот и биогенных аминов. Амины при этом превращаются в альдегиды или кетоны, аминокислоты — в кетокислоты и выделяется аммиак. Так, глутаматдегидрогеназа (L-глутамат:НАД(Ф) — оксидоредуктаза (дезаминирующая), КФ 1.4.1.3) принимает участие в следующем превращении глутамата:
Оксидоредуктазы, действующие на серосодержащие группы доноров, катализируют окисление тиоловых (сульфгидрильных) групп до дисульфидных, а сульфитов — до сульфатов. Примером фермента является дигидролипоилдегидрогеназа (КФ 1.8.1.4), катализирующая одну из промежуточных реакций окислительного декарбоксилирования пирувата:
Оксидоредуктазы, действующие на пероксид водорода в качестве акцептора, сравнительно немногочисленны и объединены в отдельный подкласс, известный также под тривиальным названием пероксидазы. Примером фермента является глутатионпероксидаза (глутатион:Н2О2 — оксидоредуктаза. КФ 1.11.1.9), участвующая в инактивации пероксида водорода в эритроцитах, печени и некоторых других тканях:
Оксидоредуктазы, действующие на пару доноров с включением молекулярного кислорода, или монооксигеназы — ферменты, катализирующие окисление органических соединений молекулярным кислородом, приводящее к включению одного из атомов кислорода в молекулы этих соединений. При этом второй атом кислорода включается в молекулу воды. Так реакция превращения фенилаланина в тирозин катализируется фенилаланин-4-монооксигеназой (КФ 1.14.16.1):
У некоторых людей генетический дефект этого фермента служит причиной заболевания, которое носит название фенилкетонурии. К монооксигеназам относится также фермент, известный под названием цитохром Р450 (КФ 1.14.14.1) Он содержится, главным образом, в клетках печени и осуществляет гидроксилирование чуждых организму липофильных соединений, образующихся в качестве побочных продуктов реакций или попадающих в организм извне. Например, индол, образующийся из триптофана в результате деятельности микроорганизмов кишечника, подвергается в печени гидроксилированию по следующей схеме:
Появление гидроксильной группы повышает гидрофильность веществ и облегчает их последующий вывод из организма. Кроме того, цитохром Р450 принимает участие в отдельных этапах превращения холестерина и стероидных гормонов. Наличие у живых организмов высокоэффективной системы цитохрома Р450 приводит в ряде случаев к нежелательным практическим следствиям: сокращает время пребывания в организме человека лекарственных препаратов и тем самым снижает их терапевтический эффект. Оксидоредуктазы, действующие на один донор с включением молекулярного кислорода, или диоксигеназы, катализируют превращения, в ходе которых оба атома молекулы О2включаются в состав окисляемого субстрата. Например, в процессе катаболизма фенилаланина и тирозина происходит образование из гомогентизиновой кислоты малеилацетоацетата, в состав которого включаются оба атома кислорода:
Фермент, катализирующий эту реакцию, называется гомогентизат-1,2-диоксигеназой (КФ 1.13.11.5). В ряде случаев встречается врождённый дефицит этого фермента, что приводит к развитию заболевания, называемого алкаптонурией. |
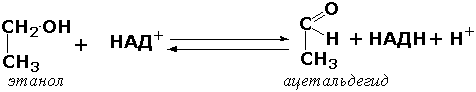
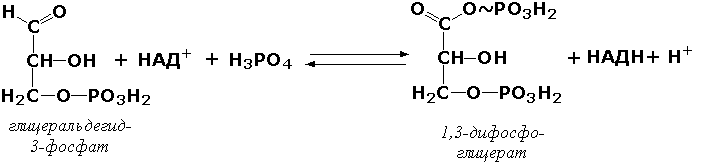


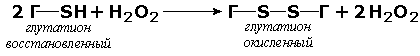

| Раздел 7.6.2 | ТРАНСФЕРАЗЫ. |
| | |
| Трансферазы — класс ферментов, катализирующих перенос функциональных групп от одного соединения к другому. В общем виде эти превращения можно записать:
где Х — переносимая функциональная группа. AX — донор группировки, В — акцептор. Подразделение на подклассы зависит от природы переносимых группировок. Трансферазы, переносящие одноуглеродные фрагменты. К этому подклассу относятся ферменты, ускоряющие перенос метильных (—CH3), метиленовых (—СН2—), метенильных (—СН=), формильных и родственных им групп. Так, при участии гуанидинацетат-метилтрансферазы (S-аденозилметионингуанидинацетат-метилтрансфераза, КФ 2.1.1.2) происходит синтез биологически активного вещества креатина:
Трансферазы, переносящие остатки карбоновых кислот (ацилтрансферазы). Они катализируют разнообразные химические процессы связанные с переносом остатков различных кислот (уксусной, пальмитиновой и др.) преимущественно от тиоэфиров коэнзима А на различные акцепторы. Примером реакции трансацетилирования может быть образование медиатора ацетилхолина при участии холин-ацетилтрансферазы (ацетил-КоА:холин-О-ацетилтрансфераза, КФ 2.3.1.6):
Трансферазы, переносящие гликозильные остатки (гликозилтрансфсразы), катализируют транспорт гликозильных остатков из молекул фосфорных эфиров к молекулам моносахаридов, полисахаридов и других веществ. Эти ферменты, в частности, играют основную роль в синтезе гликогена и крахмала, а также в первой фазе их деструкции. Ещё один фермент этого подкласса — УДФ-глюкуронилтрансфераза (УДФ-глюкуронат-глюкуронил-трансфераза (неспецифичная к акцептору), КФ 2.4.1.17) — участвует в процессах обезвреживания эндогенных и чужеродных токсических веществ в печени:
Трансферазы, переносящие азотистые группы. В этот подкласс входят аминотрансферазы, ускоряющие перенос α-аминогруппы аминокислот к α-углеродному атому кетокислот. Наиболее важным из этих ферментов является аланинаминотрансфераза (L-аланин:2-оксоглутарат-аминотрансфераза, КФ 2.6.1.2). катализирующая реакцию:
Трансферазы, переносящие фосфатные группы (фосфотрансферазы). Эта группа ферментов катализирует биохимические процессы, связанные с транспортом остатков фосфорной кислоты на различные субстраты. Указанные процессы имеют важное значение для жизнедеятельности организма, так как обеспечивают превращение ряда веществ в органические фосфоэфиры, обладающие высокой химической активностью и легко вступающие в последующие реакции. Фосфотрансферазы, использующие в качестве донора фосфата АТФ, принято называть киназами. Широко распространённым ферментом является гексокиназа (ATФ:D-гексоза-6-фосфотрансфераза. КФ 2.7.1.1.), ускоряющая перенос фосфатной группы с АТФ на моносахариды:
В некоторых случаях возможен и обратный перенос фосфатной группы с субстрата на АДФ с образованием АТФ. Так, фермент фосфоглицераткиназа (АТФ:D-3-фосфоглицерат-1-фосфотрансфераза, КФ 2.7.2.3) осуществляет превращение упомянутого ранее (см. «Оксидоредуктазы») 1.3-дифосфоглицерата:
Подобные реакции фосфорилирования АДФ с образованием АТФ, сопряжённые с превращением субстрата (а не с переносом электронов в дыхательной цепи), получили название реакций субстратного фосфорилирования. Роль этих реакций в клетке значительно возрастает при недостатке кислорода в тканях. |



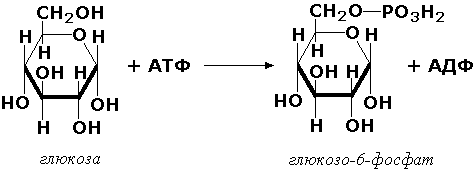

| Раздел 7.6.3 | ГИДРОЛАЗЫ. |
| | |
| Гидролазы — класс ферментов, катализирующих реакции расщепления органических соединений при участии воды (реакции гидролиза). Эти реакции протекают по следующей схеме:
где А-В — сложное соединение, А-Н и В-ОН — продукты его гидролиза. Реакции этого типа активно протекают в организме; они идут с выделением энергии и, как правило, необратимы. Подклассы гидролаз формируются в зависимости от типа гидролизуемой связи. Наиболее важными являются следующие подклассы: Гидролазы, действующие на сложные эфиры (или эстеразы) гидролизуют сложные эфиры карбоновой, фосфорной, серной и других кислот. Широко распространённым ферментом этого подкласса является триацилглицероллипаза (гидролаза эфиров глицерола, КФ 3.1.1.3). ускоряющая гидролиз ацилглицеролов:
Другие представители эстераз расщепляют сложноэфирные связи в ацетилхолине (ацетилхолинэстераза), фосфолипидах (фосфолипазы), нуклеиновых кислотах (нуклеазы), фосфоорганических эфирах (фосфатазы). Гидролазы, действующие на гликозидные связи (гликозидазы) ускоряют реакции гидролиза олиго- и полисахаридов, а также других соединений, содержащих моносахаридные остатки (например, нуклеозидов). Характерным представителем является сахараза (β-D-фруктофуранозид-фруктогидролаза, КФ 3.2.1.26). катализирующая расщепление сахарозы:
Гидролазы, действующие на пептидные связи (пептидазы), катализируют реакции гидролиза пептидных связей в белках и пептидах. К этой группе относятся пепсин, трипсин, химотрипсин, катепсин и другие протеолитические ферменты. Гидролиз пептидных связей происходит по следующей схеме:
Гидролазы, действующие на C-N-связи, отличающиеся от пептидных, — ферменты, ускоряющие гидролиз амидов органических кислот. Представитель этого подкласса — глутаминаза(L-глутамил-амидогидролаза, КФ 3.5.1.2) — участвует в поддержании кислотно-основного состояния организма, катализируя в почках гидролиз глутамина:
|
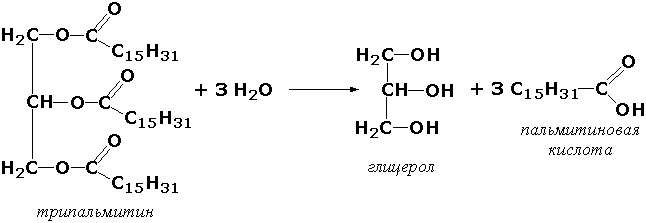
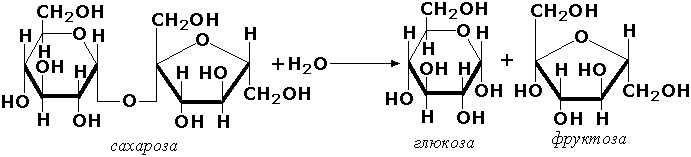
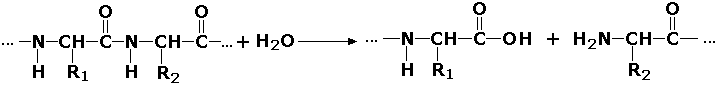

| Раздел 7.6.4 | ЛИАЗЫ. |
| | |
| Лиазы — класс ферментов, катализирующих негидролитические реакции расщепления субстратов с образованием двойных связей или, наоборот, присоединения по месту разрыва двойной связи. Общая схема этих реакций:
где А—В — субстрат, А и В — продукты реакции. В результате таких реакций часто выделяются простые вещества, например, СО2, NH3, H2О. Углерод-углерод-лиазы катализируют разрыв связи между двумя атомами углерода. Среди них наибольшее значение имеют карбокси-лиазы (декарбоксилазы), под влиянием которых осуществляется декарбоксилирование a-кето- и аминокислот, лиазы кетокислот , к которым относится цитратсинтаза, альдегид-лиазы (альдолазы). К последним относитсяфруктозодифосфатальдолаза (фруктозо-1,6-дифосфат-D-глицеральдегид-3-фосфат-лиаза, КФ 4.1.2.13), катализирующая реакцию:
Углерод-кислород-лиазы катализируют разрыв связи между атомами углерода и кислорода. В этот подкласс входят прежде всего гидро-лиазы, участвующие в реакциях дегидратации и гидратации. Примером может служить сериндегидратаза (L-серин-гидро-лиаза (дезаминирующая), КФ 4.2.1.3), осуществляющая превращение:
Иногда за основу рабочего названия может быть принята обратная реакция с применением термина «гидратаза». Так, для фермента цикла трикарбоновых кислот L-малат-гидро-лиазы (КФ 4.2.1.2) рекомендовано название «фумаратгидратаза»:
Углерод-азот-лиазы участвуют в отщеплении азотсодержащих групп. Представителем этого подкласса является гистидин-аммиак-лиаза (L-гистидин-аммиак-лиаза, КФ 4.3.1.3), участвующая в дезаминировании гистидина:
Углерод-сера-лиазы катализируют отщепление сульфгидрильных групп. К этому подклассу относятся десульфгидразы серу содержащих аминокислот, например,цистеиндесульфгидраза (L-цистеин-сероводород-лиаза (дезаминирующая), КФ 4,4.1.1). |

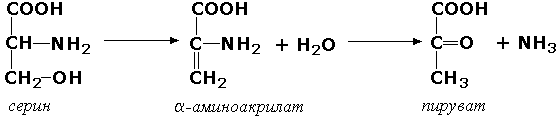
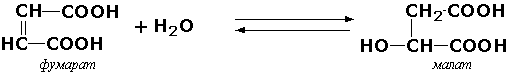
| Раздел 7.6.5 | ИЗОМЕРАЗЫ. |
| | |
| Изомеразы — класс ферментов, ускоряющих процессы внутримолекулярных превращений с образованием изомеров. Схематически реакции такого типа могут быть представлены следующим образом:
где А и А’ — вещества-изомеры. Изомеразы — сравнительно немногочисленный класс ферментов, он подразделяется на следующие подклассы в зависимости от типа катализируемой реакции изомеризации: Рацемазы и эпимеразы катализируют взаимопревращение изомеров, содержащих асимметрические атомы углерода. Рацемазами называются ферменты, действующие на субстраты с одним асимметрическим атомом, например, превращающие L-аминокислоты в D-аминокислоты. Одним из таких ферментов является aлaнинрацемаза (аланин-рацемаза. КФ 5.1.1.1), катализирующая реакцию:
Эпимеразами называются ферменты, действующие на субстраты с несколькими асимметрическими атомами углерода. К таким ферментам относится УДФ-глюкозо-эпимераза (УДФ-глюкоза-4-эпимераза, КФ 5.1.3.2). участвующая в процессах взаимопревращения моносахаридов:
Цис-транс-изомеразы — ферменты, вызывающие изменение геометрической конфигурации относительно двойной связи. Примером такого фермента являетсямалеилацетоацетатизомераза (малеилацетоацетат-цис-транс-изомераза, КФ 5.2.1.2), участвующая в катаболизме фенилаланина и тирозина и переводящая малеилацетоацетат (см. 4.6.1) в фумарилацетоацетат:
Внутримолекулярные оксидоредуктазы — изомеразы, катализирующие взаимопревращения альдоз и кетоз. При этом происходит окисление СН-ОН-группы с одновременным восстановлением соседней С=О-группы. Так, триозофосфатизомераза (D-глицеральдегид-3-фосфат-кетол-изомераза, КФ 5.3.1.1) катализирует одну из реакций углеводного обмена:
К изомеразам относятся также внутримолекулярные трансферазы, осуществляющие перенос одной группы с одной части молекулы субстрата на другую часть той же молекулы, ивнутримолекулярные лиазы, катализирующие реакции дециклизации, а также превращения одного типа кольца в другой. Следует подчеркнуть, что не все биохимические процессы. результатом которых является изомеризация, катализируются изомеразами. Так, изомеризация лимонной кислоты в изопимонную происходит при участии фермента аконитатгидратазы (цитрат (изоцитрат)-гидро-лиаза, КФ 4.2.1.3), катализирующей реакции дегидратации-гидратации с промежуточным образованием цис-аконитовой кислоты:
|
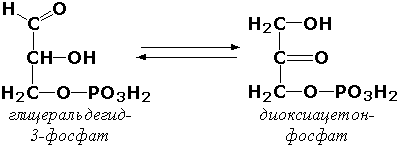
| Раздел 7.6.6 | ЛИГАЗЫ. |
| | |
| Лигазы — класс ферментов, катализирующих синтез органических соединений из активированных за счет распада АТФ (или ГТФ, УТФ, ЦТФ) исходных веществ. Для ферментов этого класса сохраняется также тривиальное название синтетазы. В связи с этим, согласно рекомендациям IUBMB, термин «синтетазы» не должен применяться для ферментов, в действии которых не участвуют нуклеозидтрифосфаты. Реакции, катализируемые лигазами (синтетазами), протекают по схеме:
где А и В — взаимодействующие вещества; А—В — вещество, образующееся в результате взаимодействия. Поскольку в результате действия этих ферментов образуются новые химические связи, подклассы VI класса формируются в зависимости от характера вновь образованных связей. Лигазы, образующие связи углерод-кислород. К ним относится группа ферментов, известных как аминокислота-тРНК-лигазы (аминоацил-тРНК-синтетазы). которые катализируют реакции взаимодействия аминокислот и соответствующих транспортных РНК. В этих реакциях образуются активные формы аминокислот, способные участвовать в процессе синтеза белка на рибосомах. Примером фермента может служить тирозил-тРНК-синтетаза (L-тирозин:тРНК-лигаза (АМФ-образующая), КФ 6.1.1.1), участвующая в реакции:
Лигазы, образующие связи углерод-сера. Этот подкласс представлен в первую очередь ферментами, катализирующими образование тиоэфиров жирных кислот с коэнзимом А. При участии этих ферментов синтезируются ацил-КоА — активные формы жирных кислот, способные вступать в различные реакции биосинтеза и распада. Рассмотрим одну из реакций активации жирных кислот, протекающую в присутствии фермента ацил-КоА-синтетазы (карбоновая кислота:коэнзим А-лигаза (АМФ-образующая). КФ 6.2.1.2):
Лигазы, образующие связи углерод-азот, катализируют многочисленные реакции введения азотсодержащих групп в органические соединения. Примером может служитьглутаминсинтетаза (L-глутамин:аммиак-γ-лигаза (АДФ-образующая), КФ 6.3.1.2). участвующая в обезвреживании токсичного продукта обмена — аммиака — в реакции с глутаминовой кислотой:
Лигазы, образующие связи углерод-углерод. Из этих ферментов наиболее изучены карбоксилазы, обеспечивающие карбоксилирование ряда соединений, в результате чего происходит удлиннение углеродных цепей. Важнейшим представителем данного класса является пируваткарбоксилаза (пируват:СО2-лигаза (АДФ-образующая), КФ 6.4.1.1), ускоряющая реакцию образования оксалоацетата — ключевого соединения цикла трикарбоновых кислот и биосинтеза углеводов:
Напомним, что реакции с участием АТФ катализируются не только ферментами VI класса, но и некоторыми ферментами II класса (фосфотрансферазами или киназами). Важно уметь отличать эти типы реакций. Их различие заключается в том, что в трансферазных реакциях АТФ является донором фосфатных групп, поэтому в результате этих реакцию не происходит выделения Н3РО4 (примеры см. выше). Наоборот, в синтетазных реакциях АТФ служит источником энергии, выделяемой при её гидролизе, поэтому одним из продуктов такой реакции будет являться неорганический орто- или пирофосфат. |
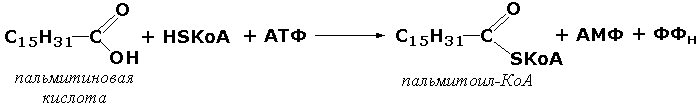
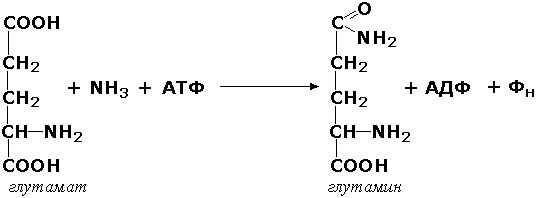
| Раздел 7.7 | Определение активности ферментов в биологическом материале. |
| Раздел 7.7.1 | Правила работы с ферментами |
| | |
| Ферменты, как все белки, являются относительно неустойчивыми веществами. Они легко подвергаются денатурации и инактивации. Поэтому при работе с ними необходимо выполнять определенные условия.
Для широкого внедрения различных биохимических (ферментативных) реакций вводится автоматизация наиболее общепризнанных и необходимых анализов, а также унификация и стандартизация лабораторных тестов. Это рационально и необходимо как для повышения точности, качества проведения проб, так и для сравнения данных, которые получены в разных лабораториях. Общепринятым является и обязательное параллельное исследование, наряду с изучаемой патологией, физиологического контроля — группы практически здоровых для установления нормальных, физиологических колебаний. Понимая относительность понятия «нормальная величина», следует принять, что для выявления различий в патологии и оценки патологического признака, за «норму», как правило, принимается средняя арифметическая М±1σ или 2σ (при нормальном Гауссовом распределении) в зависимости от степени колебания показателя. |
| Раздел 7.7.2 | Принципы определения активности ферментов в биологическом материале. |
| | |
| 5.6.2. Уникальное свойство ферментов ускорять химические реакции может быть использовано для количественного определения содержания этих биокатализаторов в биологическом материале (тканевом экстракте, сыворотке крови и т.д.). При правильно подобранных экспериментальных условиях почти всегда существует пропорциональность между количеством фермента и скоростью катализируемой реакции, поэтому по активности фермента можно судить о количественном содержании его в исследуемой пробе. Измерение ферментативной активности основывается на сравнении скорости химической реакции в присутствии активного биокатализатора со скоростью реакции в контрольном растворе, в котором фермент отсутствует или инактивирован. Исследуемый материал помещают в инкубационную среду, где созданы оптимальные температура, рН среды, концентрации активаторов и субстратов. Одновременно осуществляют постановку контрольной пробы, в которую фермент не добавляют. Спустя некоторое время реакцию останавливают путём добавления различных реагентов (изменяющих рН среды, вызывающих денатурацию белков и т.д.) и проводят анализ проб. Для того чтобы определить скорость ферментативной реакции, необходимо знать:
Наиболее часто активность фермента оценивают по количеству образовавшегося продукта реакции. Так поступают, например, при определении активности аланинаминотрансферазы, катализирующей следующую реакцию:
Определяя содержание одного из продуктов реакции – пировиноградной кислоты – в пробе после инкубации и вычитая из этого значения количество пировиноградной кислоты в контрольной пробе (в неё исследуемый материал добавляется после инкубации), находят количество продукта реакции, образовавшегося за время инкубации. Активность фермента можно рассчитывать также исходя из количества израсходованного субстрата. В качестве примера можно привести способ определения активности α-амилазы – фермента, расщепляющего крахмал. Измерив содержание крахмала в пробе до и после инкубации и вычислив разность, находят количество субстрата, расщеплённого за время инкубации. |
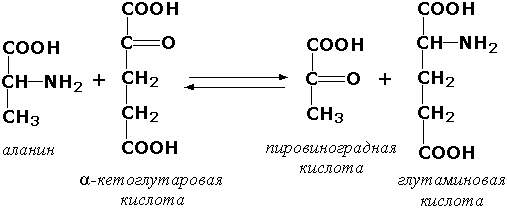
| Раздел 7.7.3 | Методы измерения активности ферментов |
| | |
| Существует большое количество методов измерения активности ферментов, различающихся по технике исполнения, специфичности, чувствительности. Чаще всего для определения применяются фотоэлектроколориметрические методы. В основе этих методов лежат цветные реакции с одним из продуктов действия ферментов. При этом интенсивность окраски получаемых растворов (измеренная на фотоэлектроколориметре) пропорциональна количеству образовавшегося продукта. Например, в процессе реакций, катализируемых аминотрансферазами, накапливаются α-кетокислоты, которые дают с 2,4-динитрофенилгидразином соединения красно-бурого цвета:
Если исследуемый биокатализатор обладает низкой специфичностью действия, то можно подобрать такой субстрат, в результате реакции с которым образуется окрашенный продукт. Примером может служить определение щелочной фосфатазы – фермента, широко распространённого в тканях человека, его активность в плазме крови существенно меняется при заболеваниях печени и костной системы. Этот фермент в щелочной среде гидролизует большую группу фосфорнокислых эфиров, как природных, так и синтетических. Одним из синтетических субстратов является паранитрофенилфосфат (бесцветный), который в щелочной среде расщепляется на ортофосфат и паранитрофенол (жёлтого цвета). За ходом реакции можно наблюдать, измеряя постепенно нарастающую интенсивность окраски раствора:
Для ферментов, обладающих высокой специфичностью действия, такой подбор субстратов, как правило, невозможен. Спектрофотометрические методы основаны на изменении ультрафиолетового спектра химических веществ, принимающих участие в реакции. Большинство соединений поглощает ультрафиолетовые лучи, причём поглощаемые длины волн характерны для присутствующих в молекулах этих веществ определённых групп атомов. Ферментативные реакции вызывают внутримолекулярные перегруппировки, в результате которых меняется ультрафиолетовый спектр. Эти изменения можно зарегистрировать на спектрофотометре. Спектрофотометрическими методами, например, определяют активность окислительно-восстановительных ферментов, содержащих в качестве коферментов НАД или НАДФ. Эти коферменты действуют как акцепторы или доноры атомов водорода и, таким образом, либо восстанавливаются, либо окисляются в процессах метаболизма. Восстановленные формы этих коферментов имеют ультрафиолетовый спектр с максимумом поглощения при 340 нм, окисленные формы этого максимума не имеют. Так, при действии лактатдегидрогеназы на молочную кислоту происходит перенос водорода на НАД, что приводит к увеличению поглощения НАДН при 340 нм. Величина этого поглощения в оптических единицах пропорциональна количеству образовавшейся восстановленной формы кофермента.
По изменению содержания восстановленной формы кофермента можно определить активность фермента. Флюориметрические методы. В основе этих методов лежит явление флюоресценции, которое заключается в том, что исследуемый объект под влиянием облучения излучает свет с более короткой длиной волны. Флюориметрические методы определения активности ферментов более чувствительны, чем спектрофотометрические. Сравнительно новыми и ещё более чувствительными являются хемилюминесцентные методы с применением люциферин-люциферазной системы. Такие методы позволяют определять скорость реакций, протекающих с образованием АТФ. При взаимодействии люциферина (карбоновой кислоты сложного строения) с АТФ образуется люцифериладенилат. Это соединение окисляется при участии фермента люциферазы, что сопровождается световой вспышкой. Измеряя интенсивность световых вспышек, удаётся определять количества АТФ порядка нескольких пикомолей (10–12 моль). Титрометрические методы. Ряд ферментативных реакций сопровождается изменением рН инкубационной смеси. Примером такого фермента является липаза поджелудочной железы. Липаза катализирует реакцию:
Образующиеся жирные кислоты могут быть оттитрованы, причём количество щёлочи, израсходованное на титрование, будет пропорционально количеству выделившихся жирных кислот и, следовательно, активности липазы. Определение активности этого фермента имеет клиническое значение. Манометрические методы основаны на измерении в закрытом реакционном сосуде объёма газа, выделившегося (или поглощённого) в ходе энзиматической реакции. С помощью таких методов были открыты и изучены реакции окислительного декарбоксилирования пировиноградной и α-кетоглутаровой кислот, протекающие с выделением СО2. В настоящее время эти методы используются редко. |
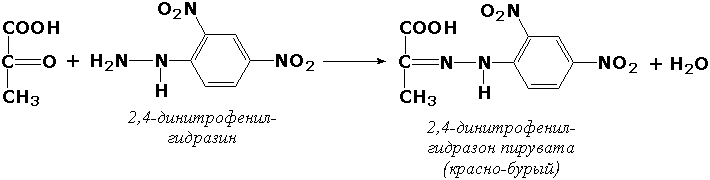
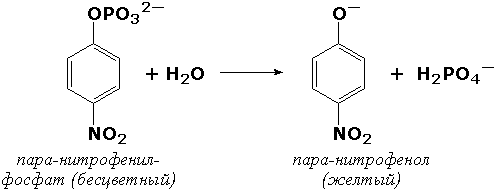


| Раздел 7.7.4 | Единицы активности ферментов и их применение. |
| | |
| Международная комиссия по ферментам предложила за единицу активности любого фермента принимать такое количество фермента, которое при заданных условиях катализирует превращение одного микромоля (10–6 моль) субстрата в единицу времени (1 мин, 1 час) или одного микроэквивалента затронутой группы в тех случаях, когда атакуется более одной группы в каждой молекуле субстрата (белки, полисахариды и другие). Должна быть указана температура, при которой проводится реакция. Результаты измерений активности ферментов могут быть выражены в единицах общей, удельной и молекулярной активности. За единицу общей активности фермента принимают такое количество фермента, которое катализирует превращение 1 мкмоль субстрата в единицу времени в расчёте на количество материала, взятого для исследования. Так, активность аланинаминотрансферазы в печени крыс равна 1670 мкмоль пирувата в час на 1 г ткани; активность холинэстеразы в сыворотке крови человека составляет 250 мкмоль уксусной кислоты в час на 1 мл сыворотки при 37°C. Особого внимания исследователя требуют высокие значения активности фермента как в норме, так и в патологии. Рекомендуется работать с небольшими показателями активности фермента. Для этого источник фермента берут в меньшем количестве (сыворотку разводят в несколько раз физиологическим раствором, а для ткани готовят меньший процентный гомогенат). По отношению к ферменту в таком случае создаются условия насыщения субстратом, что способствует проявлению его истинной активности. Общая активность фермента рассчитывается с помощью формулы:
где а – активность фермента (общая), ΔС – разность концентраций субстрата до и после инкубации; В – количество материала, взятого на анализ, t — время инкубации; n — разведение. Следует иметь в виду, что показатели активности ферментов сыворотки крови и мочи, исследуемых в диагностических целях, выражают в единицах общей активности. Поскольку ферменты являются белками, важно знать не только общую активность фермента в исследуемом материале, но и ферментативную активность белка, находящегося в данной пробе. За единицу удельной активности принимают такое количество фермента, которое катализирует превращение 1 мкмоль субстрата в единицу времени в расчёте на 1 мг белка пробы. Для вычисления удельной активности фермента необходимо общую активность разделить на содержание белка в пробе:
Например, содержание белка в ткани печени составляет 160 мг/г. Разделив общую активность аланинаминотрансферазы (см. выше) на это значение, получаем 10,4 мкмоль пирувата/мг белка × час. Чем хуже очищен фермент, тем больше в пробе находится посторонних балластных белков, тем ниже удельная активность. В ходе очистки количество таких белков уменьшается, и соответственно удельная активность фермента повышается. Предположим, в исходном биологическом материале, являющемся источником фермента (измельчённая печень, кашица из растительной ткани), удельная активность была равна 0,5 мкмоль/ (мг белка× мин). После дробного осаждения сульфатом аммония и гель-фильтрации через сефадекс она повысилась до 25 мкмоль/ (мг белка× мин), т.е. увеличилась в 50 раз. К оценке эффективности очистки ферментных препаратов прибегают при производстве лекарственных средств энзиматической природы. Удельную активность определяют в том случае, когда нужно сопоставить активность разных препаратов одного и того же фермента. Если требуется сравнить активность разных ферментов, рассчитывают молекулярную активность. Молекулярная активность (или число оборотов фермента) – это количество моль субстрата, подвергающееся превращению под действием 1 моль фермента в единицу времени (обычно в 1 минуту). Разным ферментам присуща неодинаковая молекулярная активность. Уменьшение числа оборотов ферментов происходит под действием неконкурентных ингибиторов. Изменяя конформацию каталитического центра фермента, эти вещества понижают сродство фермента к субстрату, что приводит к уменьшению числа молекул субстрата, реагирующих с одной молекулой фермента в единицу времени. |

| Примеры | Обучающие задачи и эталоны их решения. |
| | |
| 1. Задачи 1. Какие ферменты называют рацемазами? 2. Расшифруйте систематическое название фермента (отдельно для каждого из элементов, выделенных разными цветами): Определите: 2. Эталоны решения 1. Рацемазы — ферменты, катализирующие взаимопревращение оптических изомеров, содержащих единственный асимметрический атом углерода (см. раздел 2.3). 2. Систематическое название фермента читается с конца. Фермент относится к классу трансфераз, катализирует реакцию переноса метильной группы на гуанидинацетат (акцептор метильной группы) с S-аденозилметионина (донор метильной группы) (см. разделы 2.2 — 2.3). 3. а) В данной реакции происходит расщепление вещества без участия молекул воды б) Негидролитическое расщепление субстрата с образованием двух продуктов катализируют ферменты, относящиеся к четвёртому классу (лиазы) в) Разрывается связь между первым и вторым углеродными атомами, что приводит к отщеплению карбоксильной группы в виде СО2. Следовательно, подкласс фермента — углерод-углерод-лиазы (см. раздел 2.3). |